Abstracts Publicaciones 2021
Quiénes somos
Instrucciones a autores
Responsabilidad autoría
Contacto
Portal Revistas U. de Chile
Obesidad, inflamación y glucocorticoides
Obesity is a worldwide problem that also involves our country. It reduces life expectancy and represents a high economic burden for individual and the society. A large amount of economic and human resources has been allocated for obesity control and prevention. Obesity is associated with the main non-transmissible diseases on this time: cardiovascular diseases, type 2 diabetes mellitus, arterial hypertension, osteoarticular disease and some types of cancer. The development of obesity include genetic, environmental and lifestyle factors. While lipid accumulates into the adipocyte, it becomes in a tissue with hypertrophy and hyperplasia and inflammatory cells infiltrate it. Although it is not known the primary stimulus that triggers this inflammatory phenomenon, it is known this has deleterious consequences in the local and systemic metabolism and give the obese phenotype with insulin resistance, atherogenesis and inflammation. Glucocorticoids are steroidal hormones with multiple actions such as antiinflammatory properties. In obese patients, even when the plasma level of cortisol may be normal, glucocorticoids are unable to achieve regulation of the pro-inflammatory state that characterizes it. Bariatric surgery, along with recovering the weight, has been found to resolve systemic inflammatory status. The relationship between bariatric surgery, weight recovery and glucocorticoid activity has not been studied yet.
Rev Hosp Clin Univ Chile 2016; 27(4): 309-19.
Karin Papapietro V., Julia Guerrero P.
El proceso de globalización que vivimos ha provocado una serie de cambios debido a la confluencia de una compleja serie de procesos sociales, políticos, económicos y culturales que han modificado las costumbres y hábitos, pero sobre todo nuestros estilos de vida. Lo anterior ha generado un aumento considerable de las enfermedades ligadas al consumo excesivo de alimentos. Un claro ejemplo de ello es la obesidad, condición cada vez más frecuente de observar en individuos de diferentes edades y orígenes geográficos. Todos ellos comparten el hecho de consumir alimentos poco nutritivos, ricos en contenido calórico y el que realizan escasa actividad física muchas veces fomentado por las múltiples opciones que existen en los medios de transporte y a las nuevas formas de trabajo y entretenimiento de carácter más bien sedentario. Estudios han demostrado que las personas obesas desarrollan un estado de inflamación sistémica persistente donde la principal fuente de mediadores es el tejido adiposo. Interesantemente, en individuos obesos los sistemas endógenos antiinflamatorios tales como los glucocorticoides son incapaces de controlar este estado de inflamación sistémica. Revisaremos aquí la relación entre obesidad, inflamación crónica y función suprarrenal.
OBESIDAD
Tradicionalmente se ha definido la obesidad como un aumento en la proporción del tejido adiposo corporal o como un aumento patológico del tejido adiposo en relación al tejido magro. La Organización Mundial de la Salud (OMS) define la obesidad como una acumulación anormal o excesiva de grasa que puede ser perjudicial para la salud(1). Desde un punto de vista clínico se considera el índice de masa corporal (IMC), valor que relaciona peso y talla, como un método práctico y simple para realizar el diagnóstico de obesidad dado que presenta una buena correlación con la grasa corporal total. Se considera que cuando el IMC tiene un valor entre 20 y 25, el paciente está normopeso; cuando éste se sitúa entre 25 y 29,9 entonces hablamos de sobrepeso; con IMC de 30 a 34.9, obesidad grado I; obesidad grado II con IMC de 35 a 39.9 y obesidad grado III, extrema o mórbida con IMC mayor de 40(2). Existen otras formas para diagnosticar obesidad, como es la medición de los pliegues cutáneos donde el uso de ecuaciones y nomogramas permite la conversión del grosor del pliegue en grasa. Este análisis se expresa en el porcentaje de grasa corporal que varia según la edad, pero debe ser no mayor de 30% en la mujer y no mayor del 25% en el hombre, valores que se fundamentan en la consideración de que aproximadamente el 50% de la grasa corporal se encuentra en el tejido celular subcutáneo(3,4). La medición de los pliegues tiene el inconveniente de que la distribución de la grasa difiere en individuos con igual cantidad de tejido adiposo y que en ciertas formas de obesidad, la grasa tiene una distribución generalizada; mientras que en otras es fundamentalmente abdominal lo cual ha quedado demostrado en estudios que han puesto en evidencia la presencia de mayor asociación de este tipo de distribución con riesgo cardiovascular y enfermedades metabólicas. Por otra parte, la relación grasa subcutánea/grasa profunda (visceral) puede ser de 0.1 a 0.7, además de que la grasa corporal aumenta con la edad, no así el grosor del pliegue(5). También existen otros métodos para cuantificar el contenido de grasa corporal entre los que se incluyen el uso de técnicas de isótopo-dilución, de conductividad e impedancia, tomografía axial computarizada y resonancia magnética nuclear. Todas ellas son técnicas directas y precisas, pero desde el punto de vista clínico son complicadas, poco prácticas y costosas, y por ello son más utilizadas en ámbitos como la investigación(6).
La obesidad constituye un problema de salud pública que ha sido denominada la “epidemia del siglo”, a la que se ha destinado una gran cantidad de recursos económicos y humanos para su diagnóstico, control y prevención. De acuerdo con datos de la OMS, hace diez años había en el mundo aproximadamente 330 millones de adultos obesos; en 2005 esta cifra alcanzó los 400 millones de personas y en el año 2015 a lo menos 2.300 millones de individuos fueron diagnosticados con sobrepeso y más de 700 millones con obesidad(1). Si bien este problema se relacionaba como propio de los países industrializados, a la fecha el sobrepeso y la obesidad han aumentando notablemente en los países en vías de desarrollo, principalmente occidentales y en medios urbanos.
La preocupación principal de la obesidad en términos de salud pública radica en los efectos que ella tiene directamente sobre la salud y calidad de vida de las personas, y además por su fuerte asociación con las principales enfermedades no trasmisibles de nuestro tiempo: patologías cardiovasculares, diabetes mellitus tipo 2, hipertensión arterial, problemas osteoarticulares y su fuerte asociación con algunos tipos de cáncer. La obesidad puede llegar a reducir la esperanza de vida en hasta diez años y representa una elevada carga económica para el individuo y la sociedad(7-10). Chile no escapa a esta realidad: nuestro país tiene una prevalencia alta de sobrepeso y obesidad con una tendencia creciente en todas las etapas de la vida, lo que determina la existencia de aproximadamente 4 millones de personas obesas en Chile(11). Estudios del Ministerio de Salud demuestran que el sobrepeso y la obesidad representan la segunda causa de años de vida perdidos por muerte o por discapacidad prematura y la sexta causa de muerte a nivel nacional(12,13).
PATOGENIA DE LA OBESIDAD
Para desarrollar obesidad es necesario el efecto combinado de la predisposición genética y la exposición a condiciones ambientales adversas.
Los factores genéticos se relacionan con la capacidad o facilidad de acumular energía en forma de grasa tisular y una menor facilidad para liberarla en forma de calor. Así, la identificación de la mutación del gen ob en ratones genéticamente obesos ob/ob, demostró la acción de los genes en la obesidad al desarrollar estos ratones mutados obesidad, insulino-resistencia, hiperfagia y un metabolismo eficiente (engordan con la misma dieta que los ratones delgados). El gen ob es el responsable de la producción de leptina y se expresa igualmente en humanos, lo que es descrito en varias familias con obesidad temprana, acompañada de alteraciones neuroendocrinas como hipogonadismo hipogonadotrópico. Lo mismo sucede con la mutación del gen db responsable de la codificación del receptor de la leptina, mutación que también ha sido encontrada en humanos(14).
Existen otras evidencias de la participación de genes en el origen de la obesidad como son: mutaciones en el gen humano que codifica la proopiomelanocortina (POMC), condición que produce obesidad severa por falla en la síntesis de la hormona alfa- estimulante de melanocitos (a-MSH), neuropéptido que se produce en el hipotálamo e inhibe el apetito(15). A ello se suma la observación clínica que gemelos homocigotos, aun cuando crezcan separados, sus pesos siempre son parecidos y que el peso de los hijos casi siempre es parecido al de sus padres biológicos, incluso cuando hayan sido adoptados, apoyando así el papel de los genes en la etiología de la obesidad(16). A su vez, los familiares de primer grado de individuos con obesidad de comienzo en la niñez, tienen el doble de probabilidades de ser obesos que aquellos con obesidad de comienzo en la adultez(17-19). La influencia de la genética en la obesidad y la asociación de ésta a condicionantes ambientales queda de manifiesto en los numerosos reportes que muestran un incremento de los índices de obesidad en países industrializados o en vías de desarrollo en los cuales tanto la dieta rica en grasas y carbohidratos, así como los hábitos sedentarios han aumentado con el desarrollo económico(20-22).
El desarrollo de la obesidad puede ser resumido como la resultante de un aumento de la ingesta y/o una disminución del gasto energético(23). Los lípidos procedentes de la dieta o sintetizados a partir de un exceso de carbohidratos de la dieta, son transportados al tejido adiposo como quilomicrones o lipoproteínas de muy baja densidad (VLDL). Los triglicéridos de estas partículas son hidrolizados por la enzima lipoproteinlipasa localizada en los capilares endoteliales, almacenados en el tejido adiposo y reesterificados como triglicéridos tisulares. Durante los periodos de balance positivo de energía, los ácidos grasos son almacenados en las células en forma de triglicéridos; por tanto, cuando la ingestión calórica supera el gasto energético, se produce obesidad(24). En la medida en que se acumulan lípidos en los adipocitos, éstos se hipertrofian y en el momento en que la célula ha alcanzado su tamaño máximo, se forman nuevos adipocitos a partir de los preadipocitos o células precursoras y se establece así la hiperplasia de este tejido.
El tejido adiposo de los obesos posee un número aumentado de células inflamatorias, lo que se observa tanto en modelos animales de obesidad como en humanos(25). Se desconoce aún el estímulo primario que gatilla el fenómeno inflamatorio del tejido adiposo, pero la evidencia actual permite plantear que sus características son muy similares a las observadas en cualquier otro proceso inflamatorio agudo que se hace crónico, involucrando modificaciones del endotelio local las que permiten el paso selectivo de neutrófilos, macrófagos y linfocitos.
Se ha determinado que la acumulación de macrófagos en el tejido adiposo tiene consecuencias nefastas para el metabolismo local y sistémico: estudios utilizando modelos de obesidad murina en los que se evita la infiltración de macrófagos en el tejido adiposo a través de la ablación génica de diferentes moléculas proinflamatorias, mostraron que los animales no desarrollan las alteraciones metabólicas sistémicas propias de la obesidad(26,27). Por otro lado, animales de peso normal a los que se les aumenta el contenido de macrófagos en el tejido adiposo, sí desarrollan alteraciones metabólicas pese a su condición de normopeso(28). No solo las manipulaciones cuantitativas de los macrófagos tisulares repercuten en el metabolismo sistémico, también lo hacen las manipulaciones en el estado inflamatorio de estas células, aunque el número de ellas no cambie. Así, por ejemplo, la ablación selectiva de proteínas proinflamatorias en células mieloides (precursores de monocitos/ macrófagos) inducen mejoría metabólica sistémica en ratones obesos(29), mientras que cuando estas células se tornan más inflamatorias -al evitar la expresión de mediadores antiinflamatorios por parte de las células- el metabolismo sistémico empeora(30). Antecedentes recientes permiten plantear un escenario similar para la obesidad humana: un mayor contenido de macrófagos en el tejido adiposo de sujetos obesos fue el principal determinante de las alteraciones en la sensibilidad insulínica cuando se compararon sujetos obesos con o sin insulinoresistencia(31).
La acumulación local de macrófagos tiene efectos sobre la fisiología de los adipocitos y las células preadiposas, ya que factores derivados de los macrófagos y en particular el factor de necrosis tumoral alfa (TNF-a), modifican el perfil de expresión y secreción de adipoquinas por parte de las células adiposas(32). Así, el tejido adiposo inflamado se torna en un tejido que genera daño al organismo dado que condiciona el desarrollo de insulino-resistencia, aterogénesis e inflamación sistémica. Por otra parte, los productos secretados por el tejido adiposo tienen consecuencias autocrinas, sistémicas y en órganos adyancentes.
La obesidad, entonces, condiciona un estado de inflamación a nivel sistémico que se manifiesta por altos niveles en sangre de mediadores inflamatorios como son las proteínas de fase aguda tales como interleuquina (IL) 6 y la proteína C reactiva (PCR), además del TNF-a y otras interleuquinas. Los niveles plasmáticos de los mediadores inflamatorios están asociados en forma positiva con la magnitud de los depósitos adiposos valorados en forma de índice de masa corporal, porcentaje de grasa corporal, circunferencia cintura y también con las consecuencias metabólicas de la obesidad: insulino-resistencia, dislipidemia y presión arterial, tanto en adultos como en niños obesos(33,34). No hay claridad si este aumento de los mediadores inflamatorios circulantes es consecuencia de la modificación inflamatoria del tejido adiposo o si se trata de www.redclinica.cl 313 un efecto secundario a la activación de leucocitos circulantes que infiltran el tejido adiposo.
GLUCOCORTICOIDES Y CONTROL DE LA INFLAMACIÓN
Los glucocorticoides (GC) son hormonas de naturaleza esteroidal sintetizadas y liberadas por la glándula suprarrenal en respuesta a la activación del eje hipotálamo-hipófisis-suprarrenal. En los humanos, el cortisol es el GC por excelencia. Estas hormonas tienen múltiples acciones biológicas y la mayoría de sus efectos son mediados por la proteína receptora de GC (glucocorticoid receptor, GR). Entre las múltiples acciones de los GC destaca el rol antiinflamatorio(35) por lo cual ocupan un importante lugar en la farmacoterapia de enfermedades con sustrato inflamatorio(36). Dado que el cortisol es una molécula de naturaleza lipídica, su paso por vía sanguínea requiere de una proteína transportadora, rol que cumple la globulina transportadora de cortisol (cortisol binding protein, CBP)(37), pero además aproximadamente 15% del cortisol total utiliza a albúmina como transportador y solo una fracción menor del total de cortisol sanguíneo se encuentra no unido a proteínas y esta corresponde a la fracción libre de cortisol(38). En general, la acción antiinflamatoria de los GC se explica por la capacidad de inhibir la acción de moléculas asociadas a procesos inflamatorios tales como citoquinas, chemoquinas, moléculas de adhesión y por regular en forma positiva a mediadores con acción antiinflamatoria.
RECEPTOR DE GLUCOCORTICOIDES (GR)
La mayoría de las acciones celulares de cortisol requieren de la unión de éste a su receptor y de la translocación de éste desde el citoplasma al núcleo, compartimento celular donde el complejo GR/cortisol cumple un importante rol en la regulación de genes(35). GR funciona como factor de transcripción activado por ligando: en ausencia de su ligando, el GR permanece en el citoplasma formando un complejo multiproteico con proteínas chaperonas tales como inmunofilinas y heat shock proteins. Cuando se une a cortisol ocurre un cambio conformacional en GR que le permite liberarse del complejo multiproteico, exponer el sitio de localización nuclear y traslocar al núcleo en forma de complejo GR/GC y unirse a sitios específicos del DNA denominados elementos de respuesta a glucocorticoides (GRE, glucocorticoid response elements) y controlar así la transcripción de genes, efecto que se denomina transactivación (Figura 1). GR también puede unirse a otros factores de transcripción interfiriendo de forma negativa con su función, efecto denominado transrepresión. Y un tercer mecanismo de acción de GR/GC es la represión de la activación de genes por medio de su unión a sitios GRE negativos (nGRE) como ocurre para proopiomelanocortina, prolactina y osteocalcina.
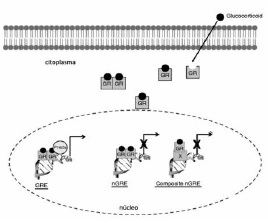
Figura 1. Mecanismos de acción de glucocorticoide. El receptor de glucocortioide (GR) es un factor nuclear residente en el citoplasma celular que es activado por la unión de su ligando.
El RNA mensajero (mRNA) del gen humano de GR tiene variantes del procesamiento alternativo (alternative splicing) del exón 9, generando las isoformas de splicing denominadas GRa si utiliza el exón 9a y GRb si utiliza el exón 9b(39,40). Ambas proteínas se diferencian en su extremo carboxilo en la cual GRa tiene una secuencia única de 50 aminoácidos, une a cortisol, interactúa con el DNA y otros factores de transcripción mientras que la isoforma GRb presenta 15 aminoácidos distintos, no une cortisol y al formar dímeros con GRa actúa como dominante negativo interfiriendo en las funciones biológicas de GRa (Figura 2). GRb ha sido identificado en células inmunes y en diversos tejidos, siempre en cuantía significativamente menor a GRa(41). Se sabe que en situaciones inflamatorias, mediadores como citoquinas favorecen el incremento de GRb y un menor efecto biológico de los GC por lo que es señalado como un mecanismo de resistencia a GC(42,43).
OBESIDAD Y GLUCOCORTICOIDES
El rol de los GC en la patogenia de la obesidad no es clara(44). Se sabe que exposiciones agudas a GC se asocian a lipólisis mientras que exposiciones crónicas y prolongadas son favorecedoras de lipogénesis. La cuantificación de los valores de cortisol sanguíneo en pacientes obesos es contradictoria: mientras algunos autores informan de valores elevados en plasma, otros autores han reportado resultados diferentes(45). La valoración de cortisol en obesos también ha sido realizada en muestras de saliva(46) y en pelo(47) y estos resultados también son contradictorios. A lo anterior se agrega el que los pacientes obesos podrían tener comprometido el metabolismo tisular del cortisol(48). Sabemos que la actividad inflamatoria mantenida, en general, se asocia con córtico-resistencia, término que hace referencia a que aun cuando exista nivel sanguíneo adecuado de la hormona, a nivel celular no existe efectividad de las funciones biológicas reguladas por ella y en este efecto podría estar involucrado el GR(43). En pacientes obesos con síndrome metabólico se ha planteado que existe un rol central de los GC en dicha patogenia por hiperactividad del eje hipotálamo-hipófisis-suprarrenal(49) donde el origen de esta activación; sin embargo, no está aclarada. Interesantemente, este incremento del cortisol es incapaz de conseguir la regulación del estado proinflamatorio que caracteriza a la obesidad. Se sabe que la expansión del tejido adiposo ocurre por incremento del número de céulas (adipogénesis) como por el incremento del tamaño de éstas (hipertrofia) y en ambos los mecanismos involucrados son complejos e incluyen la activación de una serie de factores de transcripción, entre ellos GRa. Dado que los GC tienen también un efecto promotor de la lipólisis, es posible plantear la participación de otros receptores de GC tal como GR beta cuya expresión se ha demostrado en tejido adiposo blanco de humanos(50), expresión que es regulada por condiciones metabólicas(51), por una parte, y por el exceso de cortisol, por otra(52). Estudios experimentales en modelos celulares de ratones han demostrado que la expresión de GRb incrementa por efecto de insulina sin modificación en la expresión de GRa, participando en la vía proliferativa de insulina(53); y en el modelo animal de obesidad inducida por dieta, recientemente se ha demostrado que GRb induce estatosis e inflamación hepática(54). Entonces, es posible plantear que la expresión incrementada de esta isoforma de GR tenga un rol central en el desarrollo de la resistencia a glucocorticoides de pacientes obesos. Si intervenciones farmacológicas que modifiquen la expresión y/o actividad biológica de GRb pudieran constituir un nuevo target de acción clínica, es una estrategia no explorada hasta la fecha.
Por otra parte, un estudio previo con pacientes obesos chilenos sometidos a cirugía bariátrica, ha demostrado mejoría de los parámetros inflamatorios después de un bypass gástrico, objetivándose así el control del status inflamatorio que caracteriza a la obesidad(55). A la fecha no ha sido documentado si en pacientes obesos existe resistencia a los glucocorticoides y, de existir, si ésta es reversible con la cirugía bariátrica, concordando con el control del status inflamatorio sistémico antes señalado.
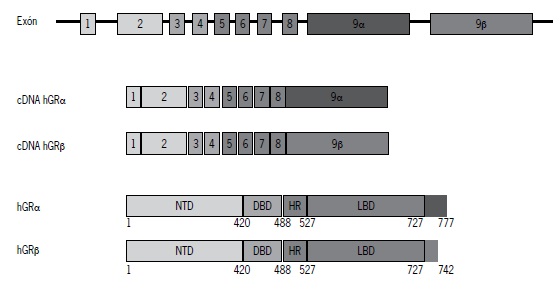
Figura 2. El procesamiento alternativo (alternative splicing) del mRNA del GR genera dos mRNA que codificarán dos proteína GR diferentes: GRa y GRb. Esta última no une ligando.
NTD: dominio N terminal; DBD: dominio de unión al DNA; HR; región bisagra; LBD: dominio de unión al ligando.
CONCLUSIONES
La obesidad es una condición clínica relevante por su asociación con patologías crónicas no transmisibles, pero con alto impacto económico dado su prevalencia en individuos laboralmente activos. En la patogenia de la obesidad, destaca el desarrollo de un estado de inflamación sistémica mantenida. Interesantemente, los mecanismos endógenos antiinflamatorios tales como el control hormonal mediado por los glucocorticoides, son incapaces de controlar en forma efectiva esta respuesta inflamatoria asociada a la obesidad. Nos parece posible pantear que en obesidad pudiera existir resistencia tisular inducida por la propia obesidad y que dificulta la acción de mecanismos fisiológicos antiinflamatorios como el mediado por los glucocorticoides.
REFERENCIAS
1. WHO. Preventing chronic diseases a vital investment. Geneve, Switzerland 2005.
2. National Health, Lung and Blood Institute Clinical Guidelines on the identifications, evaluations and treatment of overweight and obesity in adult. The evidence report. Obes Res 1998;6(suppl 2) S51-S290.
3. Monterrey Gutiérrez P, Porroto Maury C. Procedimiento gráfico para la evaluación del estado nutricional de los adultos según el IMC. Rev Cubana Aliment Nutr 2001;15:62-70.
4. Lohman TG. Skinfolds and body density and their relation to body fitness: a review. Hum Biol 1981;53:181-225.
5. Bray GA. Obesity: basic consideration and clinical approaches. Dis Mon 1989;35:449- 53.
6. Flier JS, Foster DW. Eating Disorders: Obesity, anorexia nervosa and bulimia. En: William’s textbook of Endocrinology. 9 ed Philadelphia: Sunders company;1998:1061-83.
7. Jia H, Lubetkin EI. The statewide burden of obesity, smoking, low income and chronic diseases in the United States. J Public Health (Oxf) 2009;31:496-505.
8. Finegood DT. Canada in context: challenging our epidemics of obesity and obesity-related chronic diseases. Health Rep 2009;20:9-10.
9. Moffatt E, Shack LG, Petz GJ, Sauvé JK, Hayward K, Colman R. The cost of obesity and overweight in 2005: a case study of Alberta, Canada. Can J Public Health 2011;102:144-8.
10. Colagiuri S, Lee CM, Colagiuri R, Magliano D, Shaw JE, Zimmet PZ et al. The cost of overweight and obesity in Australia. Med J Aust 2010;192:260-4.
11. Vio F, Albala C, Kain J. Nutrition transition in Chile revisited: mid-term evaluation of obesity goals for the period 2000-2010. Public Health Nutr 2008;11:405-12.
12. Ministerio de Salud. Encuesta Nacional de Salud ENS 2009-2010. disponible en: http://www.redsalud.gov.cl/portal/docs/page/ minsalcl/g_home/submenu_portada-2011/ ens2010.pdf; Ministerio de Salud. Estudio de carga de enfermedad y carga atribuible, Chile 2007.
13. Ratner R, Sabal J, Hernández P, Romero D, Atalah E. Estilos de vida y estado nutricional de trabajadores chilenos de empresas públicas y privadas de dos regiones de Chile. Rev Méd Chile 2008;136:1406-14.
14. Zhang Y. Positional cloning of the mouse obese gene and its human’s homologue. Nature 1994;372:425-32.
15. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe early onset of obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998;19:155.
16. Narkiewicz K, Szczech R, Winnicki M, Chrostowska M, Pawlowski R, Wieslawa LS et al. Heritability of plasma leptin levels: a twin study. J Hypertension 1999;117:27-31.
17. Van der Sande ME Walraven GE, Milligan PJ, Banya WA, Ceesay SM, Nyan OA et al. Family history: an opportunity for early intervention and improved control of hypertension, obesity and diabetes. Bull World Heath Organ 2001;79:321-8.
18. Farooqi IS. Genetic and hereditary aspects of childhood obesity. Best Pract Clin Endocrinol Metab 2005;19:359-74.
19. Makrilakis K, Lavranos G, Nikolaou A, Koliaki C, Doufexis D, Katsilambros N. Correlation of family history of obesity and diabetes mellitus with BMI of Greek medical students. Nutr Metab Cardiovasc Dis 2008;18:e7-8.
20. Hill JO, Peters JC. Environmental contributions of the obesity epidemic. Science 1998;280:1371-4.
21. Lindstrom J, Peltonen M, Tuomilehto J. Lifestyle strategies for weight control: experience from the Finnish Diabetes Prevention Study. Proc Nutr Soc 2005;64:81-8.
22. Popkin BM, Duffey K, Gordon-Larsen P. Environmental influences of food choice, physical activity and energy balance. Physiol Behav 2005;86:603-13.
23. Raviessin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG et al. Reduced rate of energy expenditure as a risk factor for body- weight. New Engl J Med 1988;318:467- 720.
24. Langhans W. Role of the liver in the metabolic control of eating: what we know and we do not know. Neurosci-Biobehav Rev 1996;20:145-53.
25. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin lnvest 2003;112:1796- 808.
26. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K et al. CCR2 modulates inflammatory and metabolic effects of highfat feeding. J Clin Invest 2006;116:115-24.
27. Kim DH, Sandoval D, Reed JA, Matter EK, Tolod EG, Woods SC et al. The role of GMCSF in adipose tissue inflammation. Am J Physiol Endocrinol Metab 2008;295:E1038- 46.
28. Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 2006;281:26602-14.
29. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191-8.
30. Odegaard JI, Ricardo-González RR, Goforth MH, Morel CR, Subramanian V, Mukundan L et al. Macrophage-specific PPAR gamma controls alternative activation and improves insulin resistance. Nature 2007;447:1116-20.
31. Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab 2010;299:E506-15.
32. Yamashita A, Soga Y, Iwamoto Y, Asano T, Li Y, Abiko Y et al. DNA microarray analyses of genes expressed differentially in 3T3-L1 adipocytes co-cultured with murine macrophage cell line RAW264.7 in the presence of the toll-like receptor 4 ligand bacterial endotoxin. Int J Obes (Lond) 2008;32:1725-29.
33. Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA 1999;282:2131-35.
34. Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd, Criqui M et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003;107:499- 511.
35. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids-New mechanism for old drugs. N Engl J Med 2005;353:1711-23.
36. Webster JI, Tonelli L, Stemberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol 2002;20:125-53.
37. Lin HY, Muller YA, Hammond GL. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Mol Cell Endocrinol 2010;316:3-12.
38. Lewis JG, Bagley CJ, Elder PA, Bachman AW, Torpy DJ. Plasma free cortisol fractional reflects levels of functioning corticosteroid- binding globulin. Clin Chim Acta 2005;359:189-94.
39. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potencial endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 1995;95:2435-41.
40. Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanism of action. J Biol Chem 1999;274:27857-66.
41. Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA 2001;98:6865-70.
42. Lewis-Tuffin LJ, Cidlowski JA. The physiology human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci 2006;1069:1-9.
43. Guerrero J, Gatica HA, Rodríguez M, Estay R, Goecke IA. Septic serum induces glucocorticoids resistance and modifies the expression of glucocorticoid isoforms receptors: a prospective cohort study and in vitro experimental assay. Crit Care 2013;17:R107.
44. Bjorntorp P, Rosmond R. Obesity and cortisol. Nutrition 2000;16:924-36.
45. Praveen EP, Sahoo JP, Kulshreshtha B, Khurana ML, Gupta N, Dwivedi SN et al. Morning cortisol is lower in obese individuals with normal glucose tolerance. Diabetes Metab Syndr Obes 2011;4:347–52.
46. Abraham SB, Rubino D, Sinaii N, Ramsey S, Nieman LK. Cortisol, obesity and the metabolic syndrome: a cross-sectional study of obese subjects and review of the literature. Obesity (Silver Spring) 2013;21:E105-17.
47. Olstad DL, Ball K, Wright C, Abbott G, Brown E, Turner AI. Hair cortisol levels, perceived stress and body mass index in women and children living in socioeconomically disadvantaged neighborhoods: the READI study. Stress 2016;19:158-76.
48. Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O et al. Tissuespecific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 2001;86:1418-21.
49. Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J Clin Endocrinol Metab 2009;2692-701.
50. Syed AA, Irving JA, Redfern CP, Hall AG, Unwin NC, White M et al. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity 2006;14:759-64.
51. Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM et al. Discovery of glucocorticoid receptor - b in mice with a role in metabolism. Mol Endocrinol 2010;24:1715-27.
52. John K, Marino JS, Sanchez ER, Hinds Jr TD. The glucocrtiocid receptor: cause of or cure for obesity? Am J Physiol Endocrinol Metab 2016;310:E249-E257.
53. Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor b stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem 2014;289:17885-94.
54. Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD Jr. Glucocorticoid receptor b induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisime proliferatoractivated receptor (PPAR) a. J Biol Chem 2016;291:25776-88.
55. Rojas P, Carrasco F, Codoceo J, Inostroza J, Basfi-fer K, Papapietro K et al. Trace element status and inflammation parameters after 6 month of Roux -en-Y gastric bypass. Obes Surg 2011;21:561-8.
Correspondencia:
Dra. Julia Guerrero Peralta
Laboratorio de Inmunomodulación Neuroendocrina, Facultad de Medicina, U. de Chile
[email protected]
562 2297 8038