Abstracts Publicaciones 2021
Quiénes somos
Instrucciones a autores
Responsabilidad autoría
Contacto
Portal Revistas U. de Chile
Síndrome de Miller-Fisher en edad pediátrica: un debut inusual
Miller-Fisher syndrome is a peripheral neuropathy characterized by ophthalmoplegia, areflexia and ataxia. The incidence of this syndrome is estimated at 1 in a million, and just 14% of these occur on a pediatric age, making it an infrequent diagnosis in pediatrics. In this article we report a case of Miller-Fisher syndrome with an unusual onset on a pediatric patient along with infrequent radiological findings.
Rev Hosp Clín Univ Chile 2017; 28: 202 - 8
Álvaro Fischer B., Aldo Ghisoni S., María Elena Herrera G., Mariela Muñoz P., Catalina Culcay A.
El síndrome de Miller-Fisher (MFS) corresponde a una neuropatía periférica caracterizada por su tríada clásica: oftalmoplejia, ataxia y arreflexia. Fisiopatológicamente se enmarca dentro del síndrome Anti-GQ1b, en el que la infección por microorganismos portadores de epítopes lipopolisacáridos como Campylobacter jejuni induce la producción de anticuerpos IgG contra antígenos similares presentes en el ser humano, como los gangliósidos presentes en el III y IV par craneanos. Esta reacción autoinmune genera una desmielinización nerviosa periférica con un amplio espectro de manifestaciones clínicas que variarán desde la oftalmoparesia aguda hasta formas de compromiso extenso como el síndrome de Guillain-Barré (SG-B) y su variante el MFS(1–3).
La prevalencia de este síndrome en edad pediátrica es baja en comparación a la edad adulta, representando cerca del 14%(4,5). Corresponde a una variante del SG-B y se estima presente en entre un 1-10% de los niños con SG-B(6); otros estudios entregan cifras más acotadas de un 5 a 7%(4–7). Por otro lado, la recurrencia en población pediátrica es extremadamente rara, existiendo pocos reportes de casos aislados(8).
En este trabajo presentamos el caso de un escolar de 11 años, sin antecedentes mórbidos, con diagnóstico final de MFS, el que su presentación solapada significó un desafío diagnóstico. Este caso destaca, pues el paciente presentó una recurrencia un mes después de iniciado el tratamiento, siendo el cuarto caso descrito en la literatura mundial de un MFS recurrente en edad pediátrica (8).
REPORTE DE CASO
Escolar de 11 años con historia previa de sobrepeso y un cuadro diarreico agudo y fiebre de tres días de duración resuelto durante la semana previa a su ingreso, sin otros antecedentes. Consulta por cuadro de dos días de evolución de diplopía y visión borrosa de comienzo súbito, agregándose cefalea pulsátil de moderada intensidad y predominio frontal, mareos intermitentes y fotofobia. Consulta en extrasistema, donde es derivado a nuestro centro.
Ingresa al Servicio de Urgencia donde es evaluado por neurocirujano. Al examen neurológico el paciente se encuentra vigil y orientado temporoespacialmente. Destaca marcha atáxica, con aumento de la base de sustentación, pruebas de dismetría y disdiadococinesia alteradas. Se objetiva una parálisis del sexto par craneal bilateral. Reflejos osteotendíneos (ROT) conservados. Se solicitan exámenes: hemograma, electrolitos plasmáticos, calcio, fósforo, perfil bioquímico, nitrógeno ureico, gases en sangre venosa, perfil hepático y pruebas de coagulación dentro de rangos normales. Serología IgM para mycoplasma negativa. Se solicita resonancia magnética (RNM) de cerebro y es informada como normal. Se descarta patología neuroquirúrgica y se hospitaliza con diagnóstico de síndrome atáxico agudo con sospecha de rombencefalitis aguda viral.
Se hospitaliza para monitorización continua y continuar estudio. Al día siguiente presenta aumento de la cefalea y fotofobia, manteniéndose la diplopía, sin otros cambios. Se solicita evaluación por oftalmología donde destaca oftalmoplejia internuclear, con fondo de ojo normal. Exámenes realizados: punción lumbar (PL) con presión de apertura límite de 20 mmHg. Citoquímico y citológico normales; panel FilmArray® para estudio de meningitis y encefalitis en líquido cefalorraquídeo (LCR), el que resulta negativo para todos los patógenos estudiados.Se realiza una segunda RM cerebral el día 18 de abril, con informe que muestra dilatación bilateral de la vaina del nervio óptico (Figura 1).
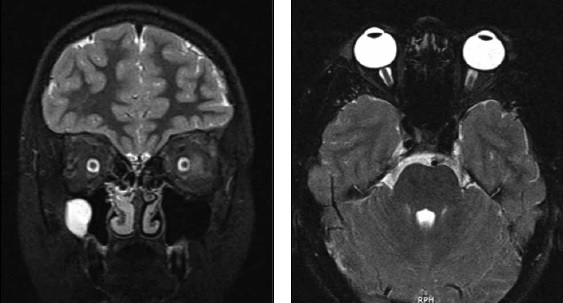
Figura 1. RMN encéfalo secuencias T2 con supresión grasa en planos coronal y axial, muestran dilatación en vaina de nervios ópticos con señal de líquido cefalorraquídeo y con aplanamiento de discos ópticos.
El segundo día de hospitalización, dada presión de apertura de LCR límite y engrosamiento de las vainas de los nervios ópticos en la RM, se propone un diagnóstico presuntivo de hipertensión intracraneana idiopática (HII). Se inicia manejo con acetazolamida y prednisona. En los días posteriores se objetiva tendencia a la normalización de las pruebas de dismetría y disdiadococinesia.
Al sexto día de hospitalización el paciente refiere ausencia de fotofobia y cefalea, con persistencia de diplopía. Al examen neurológico existe presencia de oftalmoparesia y ptosis palpebral bilateral sin paresia facial, con pupilas isocóricas, pero hiporreactivas. Temblor leve en pruebas de dismetría, marcha levemente atáxica, prueba de pequeña paresia negativa. Arreflexia rotuliana y aquiliana con fuerza y sensibilidad de extremidades sin alteraciones. Se sospecha un MFS. Se inicia terapia con inmunoglobulina 400 mg/kg/día por cinco días (dosis total de 2 gr/kg) y se suspende el tratamiento con acetazolamida y prednisona.
Al séptimo día de hospitalización destaca leve mejoría del cuadro con esbozada excursión ocular a superior bilateral, ROT esbozados, logrando sedestación. Se realiza una segunda PL, con estudio de LCR que muestra disociación albúmino citológica con proteínas de 69 mg/dl y 5 leucocitos/mm3.
Al noveno día el paciente logra excursión ocular hacia cefálico de aproximadamente 15 grados, con regresión de la ptosis y dismetría, reflejo fotomotor presente, directo y consensual, con pupilas isocóricas y simétricas. Se realizó estudio electrofisiológico que muestra ausencia de representación electrofisiológica del arco reflejo S1 bilateral, lo que en el contexto de neuroconducción sensitiva y motora normal, es compatible con MFS.
Evoluciona en su décimo primer día de hospitalización, concluido el quinto pulso de inmunoglobulinas con excursión ocular de 20 grados hacia cefálico, ataxia mínima y ROT disminuidos. Dismetría y disdiadococinesia escasas y logrando marcha en tándem, decidiéndose su alta. El paciente egresa con prednisona oral en dosis decrecientes por un mes. Dada lentitud en la recuperación oculomotora, fueron suministradas vitaminas de complejo B para optimizar la recuperación de la neuropatía.
Progresando gradualmente hacia la mejoría, reingresa 2 meses después por un cuadro de diplopía y desviación del ojo derecho. Evaluado por oftalmólogo se pesquisa estrabismo convergente derecho de 12 grados y paresia del VI par derecho, reflejos pupilares normales, papilas sin edema. Se hospitaliza y recibe gammaglobulina 2 gr/kg en 48 hrs. RM cerebral con contraste y difusión no muestra signos sugerentes de enfermedad desmielinizante ni otras alteraciones. Electromiografía sin alteraciones específicas que no muestra elementos categóricos de un defecto en la unión neuromuscular.
El paciente es dado de alta con remisión parcial de la signología neurológica. En control neurológico ambulatorio posterior evoluciona asintomático con recuperación de los movimientos oculares, sin ataxia y recuperación casi completa de los ROT.
DISCUSIÓN
El MFS, variante usual del SG-B en el adulto, es una entidad rara en pediatría, que plantea un importante desafío diagnóstico por su baja frecuencia, variable presentación y desarrollo tardío de su triada clásica. La presentación de un MFS recurrente es algo aún más infrecuente, describiéndose hasta la fecha 29 casos, de los cuales sólo dos son casos pediátricos(8).
En la historia clínica del SMF, es importante el antecedente de una infección digestiva o respiratoria una o dos semanas previo al inicio de la sintomalogía neurológica, a la que rara vez se asocia de manera aguda. Se describe en la literatura que una infección gastrointestinal se presenta en un 25% de los casos, destacando como agente Campylobacter jejuni, aunque también se asocia con otros agentes infecciosos como C. burnetti, M. pneumoniae y el virus Epstein-Barr(1,4,5,9,10). En este caso clínico existe el antecedente de un cuadro diarreico agudo siete días previo al debut, ya resuelto al momento de consultar.
La presentación de la sintomatología del MFS es variable, pudiendo ser súbita o progresiva, y presentando desde la tríada clásica hasta un cuadro generalizado de disfagia, disfonía e insuficiencia respiratoria(1). Según un estudio que incluyó a 466 pacientes con MFS, el síntoma inicial más frecuente es la alteración de la marcha (47%), seguida por diplopía (32%) y disistesias (13%). Los síntomas más descritos en el transcurso de la enfermedad son la oftalmoplejia, ataxia y arreflexia, presentes en todos los pacientes de esta serie, siendo la arreflexia el síntoma que puede aparecer más tardíamente. El promedio de instalación de los síntomas es de 4 días, describiéndose un margen de 1 a 20 días para alcanzar el nadir de la enfermedad(9).
Nuestro paciente debutó con un cuadro de cefalea, fotofobia, diplopía y un síndrome cerebeloso que motivaron un estudio inicial orientado a la búsqueda patología aguda vascular, infecciosa y tóxico- metabólica. Estos síntomas, aunque presentes en algunas descripciones realizadas por Fisher, son poco usuales dentro de las series más numerosas de MFS y plantearon una dificultad diagnóstica inicial. Sólo al desaparecer dichos síntomas y aparecer de forma gradual la tríada clásica al sexto día de hospitalización, se pudo configurar un cuadro sugerente de MFS(9,11).
El MFS presenta en un 90% de los casos anticuerpos tipo IgG contra gangliósido anti-GQ1b(12), por lo que un resultado positivo apoya certeramente el diagnóstico; sin embargo, hay otros parámetros de laboratorio concordantes con MFS como la disociación albumino citológica en LCR que puede aparecer tanto al inicio del cuadro como en la segunda o tercera semana de evolución y que se encuentra presente en un 65% de los casos. Estudios electrofisiológicos muestran un patrón de desmielinización en las vías nerviosas afectadas(13). La confirmación de conversión serológica con presencia de anticuerpos GQ1b no se encuentra disponible como opción diagnóstica en nuestro medio; sin embargo, nuestro paciente sí presentó una disociación albúmino citológica en una segunda PL realizada durante la segunda semana de evolución, lo que concuerda con el patrón clásico del MFS(1,14).
En el MFS se describe de forma frecuente un compromiso del III y IV par craneal en estudio imagenológico del MFS mediante RM, aunque el V, VII, XI y XII par pueden verse involucrados (Figura 2). Se ha descrito en la literatura el engrosamiento del II par craneano en casos pediátricos de MFS, aunque este evento resulta raro(5).
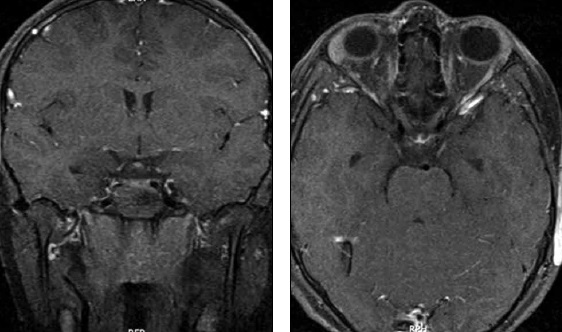
Figura 2. RNM encéfalo secuencias T1 con supresión grasa y gadolinio en plano coronal y axial donde se observa realce de ambos terceros pares.
En el caso de nuestro paciente, un informe señala que la vaina de ambos nervios ópticos se encontraba engrosada en la RM, lo que sustentaba un diagnóstico diferencial con la HII, entidad que además explicaba muchas características del cuadro del paciente, tales como diplopía, cefalea y parálisis del nervio abducens. Aunque la HII requiere de otros elementos clínicos, como presión endocraneana elevada, papiledema al fondo de ojo y una composición de LCR normal para certificar su diagnóstico(15), en este caso los hallazgos imagenológicos excepcionales y el análisis de LCR normal inicial hicieron considerar a la HII como un diferencial antes de la aparición de la tríada clásica del MFS.
El MFS es una enfermedad generalmente benigna y autolimitada; la mayoría de los pacientes resuelve 206 Revista Hospital Clínico Universidad de Chile completamente sus síntomas entre 8 y 12 semanas sin tratamiento(12). El uso de inmunoglobulinas no afecta el pronóstico de los pacientes con MFS, pero sí podría acelerar la desaparición de los síntomas(16). En el presente caso el paciente fue tratado con inmunoglobulinas y se observó el inicio de la remisión de los síntomas en las siguientes 24 horas.
La recurrencia del MFS es muy poco frecuente, especialmente en edad pediátrica, siendo este el tercer caso descrito en la literatura. Según lo observado en población adulta, la recurrencia también va precedida de un cuadro viral y presenta características clínicas similares al primer episodio, aunque a menudo más agresivas. El MFS rara vez recurre más de 2 veces en la vida, fluctuando la aparición del segundo episodio entre 18 meses y 5 años desde el debut de la enfermedad(8).
Dada la posibilidad de aparición solapada de la sintomatología del MFS y la tardía alteración del análisis del LCR, debemos destacar la importancia de una constante reevaluación y replanteamiento del enfoque diagnóstico en pacientes con sintomatología neurológica de esta naturaleza para poder iniciar el tratamiento oportunamente.
CONCLUSIÓN
El diagnóstico del MFS en pacientes pediátricos es siempre un desafío diagnóstico, principalmente por su escasa prevalencia en esta población(8). El diagnóstico es fundamentalmente clínico, apoyado por algunos exámenes de laboratorio, siendo importante descartar otras patologías con una forma de presentación similar como aquellas de índole tóxica, infecciosa, postinfecciosa y neoplásica. El caso presentado nos muestra también la amplia gama de síntomas iniciales con que se puede presentar este síndrome antes de mostrar su tríada clásica, siendo muchas veces progresiva la aparición de esta. Particularmente la cefalea y fotofobia pre sentada en este caso clínico son síntomas atípicos y no constatados en grandes estudios de cohorte de MFS(9).
Asimismo, el MFS se trata de una enfermedad de curso usualmente benigno y con buena respuesta a terapia con inmunoglobulinas que, dada la aparición tardía de su tríada diagnóstica y alteraciones en el LCR, debe tenerse permanentemente en cuenta como diagnóstico diferencial en un cuadro larvado que presenta oftalmoplejia, ataxia y/o arreflexia en ausencia de otras causas(9,16).
REFERENCIAS
1. Ostia Garza P, Fuentes Cuevas M. Síndrome de Guillain-Barré variedad Miller- Fisher. Reporte de un caso. Arch Inv Mat Inf 2011;3:30–353.
2. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013;84:576–83.
3. Uribe R, Suárez F, Sandoval P, Mellado P. Síndrome anti-GQ1b: Descripción de cuatro pacientes y revisión de la literatura. Rev Med Chil 2013;141:1211–5.
4. Berlit P, Rakicky J. The Miller Fisher syndrome. Review of the literature. J Clin Neuroophthalmol 1992;12:57–63.
5. Malhotra A, Zhang M, Wu X, Jindal S, Durand D, Makhani N. MRI findings of optic pathway involvement in Miller Fisher syndrome in 3 pediatric patients and a review of the literature. J Clin Neurosci 2017;10–4.
6. Lin JJ, Hsia SH, Wang HS, Lyu RK, Chou ML, Hung PC et al. Clinical variants of Guillain-Barré syndrome in children. Pediatr Neurol 2012;47:91–6.
7. Bassal F, Lupo P. Case 3: Ophthalmoplegia and unsteady gait in an 11-year-old boy. Pediatr Rev 2018;39:39.
8. Grosso S, Verrotti A, Tei M, Cornacchione S, Giannini F, Balestri P. Recurrent Miller Fisher syndrome in children. Pediatr Neurol 2014;50:269–71.
9. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K et al. Bickerstaff’s brainstem encephalitis and Fisher syndrome form a continuous spectrum: Clinical analysis of 581 cases. J Neurol 2008;255:674–82.
10. Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barré and Fisher syndromes. Lancet Infect Dis 2001;1:29–37.
11. Miller Fisher C. An unusual variant of acute idiopathic polyneuritis (Syndrome of ophtalmoplegia, ataxia and areflexia). N Engl J Med 1956;255:57–65.
12. Muñiz AE. Multiple cranial nerve neuropathies, ataxia and, areflexia: Miller Fisher syndrome in a child and review. Am J Emerg Med 2017;35:661.e1-661.e413.
13. Tan CY, Yuki N, Shahrizaila N. Delayed facial palsy in Miller Fisher syndrome. J Neurol Sci [Internet] 2015;358:409–12.
14. Torricelli RE. Sindrome De Guillain Barre en Pediatria. Med Buenos Aires 2009;69:84.
15. Friedman DI, Jacobson DM. Diagnostic criteria for idiopathic intracranial hypertension. Neurology 2002;59:1492–5.
16. Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher
Correspondencia:
Álvaro Nicolás Fischer Balada
[email protected]
569 7877 0372