Abstracts Publicaciones 2021
Quiénes somos
Instrucciones a autores
Responsabilidad autoría
Contacto
Portal Revistas U. de Chile
Síndrome de Vogt-Koyanagi-Harada en niños
Introduction: Vogt-Koyanagi-Harada (VKH) syndrome is a systemic inflammatory disease that causes chronic and bilateral granulomatous panuveitis, usually described in adults. Objectives: To describe manifestations and complications of VKH in pediatric patients. Methods: Retrospectivedescriptive study upon patients less than 14 years-old with VKH, attended from January 1985 to July 2010 in three different centers. Results: A total of 17 patients (34 eyes) were studied; 9 (53%) female. The mean age was 10.8 years-old. Among extraocular manifestations; neurological (71%), dermatological (29%) and auditive (24%) signs were observed. Ocular findings included optic-disc involvement (94%), anterior uveitis (79%), choroiditis (77%), serous retinal detachment (71%) and vitritis (71%). Initial visual acuity (VA) was ≤0.05 in 47% of cases and ≥0.6 in 12% of patients. 71% presented complications: glaucoma (20 eyes), sinechiae (10 eyes), maculopathy (6 eyes) cataract (5 eyes) and ptisis bulbi (1 eyes). 35% received only corticosteroids and 65% inmunosupressive drugs. After treatment, 6% had VA ≤0.05 and 59% ≥0.6. Ten patients (59%) recurred: 30% compromising posterior pole, and 50% recurred >3 times. Conclusions: VKH in children is infrequent. It presents with optic-disc involvement and complications of posterior pole. It requires a high degree of suspicion, quick evaluation and early treatment, which include inmunosupressive and extended corticosteroid therapy. Nevertheless, a high rate of recurrence is seen among this group of patients.
Rev Hosp Clín Univ Chile 2017; 28: 181 - 8
Víctor Velásquez R., Javiera Araya C., Fernanda Pérez V., Sergio Morales E., Esteban Fuentes G., Pablo Romero C.
El síndrome de Vogt-Koyanagi-Harada (VKH) corresponde a una enfermedad inflamatoria sistémica con gran potencial de producir ceguera si no se realiza tratamiento oportuno. Es caracterizada por una panuveítis granulomatosa bilateral, severa, crónica, asociada a una variedad de manifestaciones que se encuentran a nivel de sistema nervioso central, auditivo, ocular y cutáneo(1). Las manifestaciones oftalmológicas de este síndrome son variables, pudiendo llevar al compromiso severo de la visión. Estas cursan en cuatro fases(2), destacando la fase crónica-recurrente 182 Revista Hospital Clínico Universidad de Chile que se caracteriza por presentar episodios de exacerbación y remisión en el tiempo y en la cual se tienden a producir las complicaciones oculares(3).
La etiopatogenia y fisiopatología se desconoce, pero existe evidencia que sugiere que sería consecuencia de un proceso autoinmune mediado por linfocitos-T contra antígenos asociados a melanocitos(4). El mecanismo que gatilla la respuesta inmune es desconocido, pero existe cierta evidencia que sugiere que podría ser resultado de una injuria cutánea o posterior a una infección viral(5). En el ojo se produciría una reacción inflamatoria uveal granulomatosa, predominando linfocitos CD4+ en fases agudas y linfocitos CD8+ en fases de remisión(6).
El objetivo del tratamiento es la supresión de la inflamación intraocular que se logra a través del uso de antiinflamatorios esteroidales e inmunosupresores, que corresponde al principal factor de riesgo para el desarrollo de las complicaciones. Los factores pronósticos están vinculados principalmente al retraso o ausencia de complicaciones, los cuales están dados por la precocidad en el inicio del tratamiento, su respuesta y la duración de la fase crónica, además de manifestaciones propias de la enfermedad, como la agudeza visual al inicio de las manifestaciones clínicas, la extensión de los desprendimientos de retina en la fase aguda y la presencia de membranas neovasculares(7,8).
Se ha descrito gran variabilidad étnico y geográfica en la prevalencia de VKH(2), siendo más frecuente en Asia, América Latina y Medio Oriente(9). La prevalencia global es desconocida, describiéndose desde un 3% a un 21,08% de las uveítis según la población estudiada(9), siendo la causa más común de panuveítis en India(10). Afecta generalmente a adultos entre la segunda y quinta década de la vida con predilección por el género femenino(9).
El diagnostico de VKH es infrecuente en edad pediátrica, correspondiendo al 8.2%-16% de las uveítis por VKH en poblaciones de alta prevalencia(11,12). Se ha visto que el curso de la enfermedad ocular es más agresivo en niños que en adultos, describiéndose mayor compromiso visual debido al desarrollo de complicaciones como catarata, glaucoma y neovascularización coroidal, generadas por lo difícil y tardío del diagnóstico(13).
El espectro de manifestaciones clínicas ha sido documentado en varios estudios en población adulta; sin embargo, en la edad pediátrica, debido a su baja prevalencia, se desconoce tanto su curso clínico, como el tipo de manifestaciones y frecuencia(2,14).
El propósito de este estudio es describir las manifestaciones clínicas, principalmente oculares, aspectos epidemiológicos, complicaciones y la terapia empleada en la población pediátrica con diagnóstico de síndrome de VKH.
MATERIALES Y MÉTODOS
Este trabajo corresponde a un estudio retrospectivo descriptivo. Se analizaron 184 fichas de pacientes portadores de VKH controlados en atención de policlínico del Departamento de Úvea del Hospital del Salvador, el Servicio de Oftalmología del Hospital Clínico Universidad de Chile y de la consulta de uno de los autores, entre enero de 1985 y julio de 2010, seleccionando a aquellos pacientes de 14 años o menos.
Los parámetros evaluados en todos los pacientes fueron extraídos de los registros clínicos de ambos hospitales y consulta privada de uno de los autores, e incluyeron: la agudeza visual mejor corregida, utilizando tabla ocular de Snellen; biomicroscopía con lámpara de hendidura; evaluación del fondo de ojo con dilatación pupilar con lente Volk de 78 dioptrías y oftalmoscopía indirecta con una lente Volk de 20 dioptrías. Además se evaluó la presencia de manifestaciones neurológicas, auditivas y dermatológicas.
El diagnóstico de VKH se realiza mediante los criterios establecidos por la Sociedad Americana de Uveítis (AUS), publicados en el 2001(15).
Se analizaron los hallazgos oculares al ingreso, la agudeza visual mejor corregida según la mejor línea de tablas de Snellen (AVMC) expresada como una puntuación LogMAR y la presencia o ausencia de compromiso oftalmológico y extraocular. La biomicroscopía del segmento anterior y el examen de fondo dilatado se realizaron bajo examen directo con lupa Volkmann de 78 D (Volk Optical Inc., Mentor, Ohio, EUA) en la lámpara de hendidura. Se midió la presión intraocular con una tonometría de aplanación de Perkins (tonometría Perkins Clement-Clarke Inc., Columbus, Ohio, EE.UU.). El estado extraocular fue evaluado para especialistas en Dermatología, Neurología y Otorrinolaringología. Todos los individuos con dos o más manifestaciones de las categorías previamente descritas para VKH: oftalmológico, dermatológico, audiológico y neurológico fueron considerados clínicamente afectados(15,16). De las fichas se recogieron los datos relacionados con: manifestaciones oftalmológicas, edad de inicio y el grado de cumplimiento de los criterios diagnósticos. Además se analizó la presencia de complicaciones y la frecuencia de las diferentes complicaciones encontradas. Se evaluaron los tratamientos recibidos, el número de recidivas y el tipo de estas.
Se consideró defecto visual grave a la agudeza visual igual o menor de 20/200 (ceguera legal), de acuerdo con las definiciones actuales de la Organización Mundial de la Salud (OMS) de ceguera y discapacidad visual en la Clasificación Estadística Internacional de Enfermedades (CIE-10) de 2011(17). Todos los exámenes se realizaron de acuerdo con los principios de la Declaración de Helsinki.
RESULTADOS
De un total de 184 pacientes con diagnóstico de VKH, 17 (34 ojos) correspondieron a niños (9,2%); 8 eran hombres y 9 mujeres. La edad promedio de presentación fue 10,8 años (rango 4-14 años). En hombres fue de 9,5 años mientras que en mujeres fue de 12 años. El tiempo promedio de seguimiento fue 32,8 meses (rango 4 – 144 meses).
En relación a los hallazgos clínicos en frecuencia, se asoció al compromiso oftalmológico: el compromiso neurológico en un 71% (12 pacientes), dermatológico en un 29% (5 pacientes) y auditivo en un 24% (4 pacientes) (Tabla 1).

Tabla 1. Compromiso ocular y extraocular en pacientes pediátricos con síndrome de Vogt-Koyanagi-Harada
Dentro de los hallazgos clínicos oculares (Tabla 2), destaca el compromiso del disco óptico (94%) caracterizado por edema o hiperemia papilar. En cuanto a la agudeza visual (Tabla 3), un 47% de los ojos presentó al ingreso agudeza visual (AV) ≤ 0.05 (ceguera legal) y un 12% presentó AV ≥ 0,6. Posterior al tratamiento, un 6% presentó AV final ≤ 0.05, y un 59% de los ojos presentó AV final ≥ 0.6. Respecto al cumplimiento de los criterios diagnósticos, un 47% de los casos fueron incompletos, 29% completos, y 24% presuntos.

Tabla 2. Hallazgos clínicos oculares al ingreso en pacientes pediátricos cpn síndrome de VKH
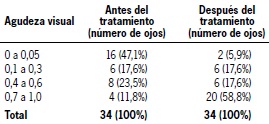
Tabla 3: Agudeza visual antes y después del tratamiento en pacientes pediátricos con síndrome de Vogt-Koyanagi-Harada
Las complicaciones se presentaron en 12 pacientes. El total de complicaciones observadas fueron 42, donde la más frecuente de estas fue glaucoma (20 ojos), seguido por sinequias (10 ojos), maculopatía (6 ojos), catarata (5 ojos) y ptisis bulbar (1 ojo).
En relación al tratamiento 35% fueron tratados únicamente con corticoides sistémicos y en un 65% se asociaron a uno o más inmunosupresores. El inmunosupresor más utilizado fue azatioprina en un 38%, luego ciclosporina en 25%, metotrexato en un 19%, ciclofosfamida en 13% y por último, un 6,3% (1 paciente) utilizó agentes biológicos (infliximab). Se registraron un total de 10 casos de recidivas (59%), donde en 70% sólo comprometió el segmento anterior del ojo, un 20% fue sólo posterior y en un 10% tuvo compromiso de ambos segmentos. La mitad de los pacientes que recidivaron presentaron más de 3 episodios.
DISCUSIÓN
El síndrome de VKH infantil corresponde a una enfermedad multisistémica crónica con escasos reportes en la literatura debido a su baja frecuencia; sin embargo, puede provocar pérdida de la visión en la niñez, ya que es un cuadro por el que se debe tener un alto grado de sospecha para un tratamiento oportuno.
En nuestra muestra de pacientes con VKH se encontró que el 9,2% correspondía a niños menores de 14 años, lo cual está dentro del rango publicado en poblaciones con alta prevalencia(9), observándose una leve predilección por el sexo femenino, al igual como señalan estudios de VHK en población general(8).
En relación a las manifestaciones extraoculares observadas, alrededor del 70% tiene manifestaciones neurológicas, 30% cutáneas y 25% auditivas, similar a lo documentado en la literatura respecto pacientes con VKH a de todas las edades(9).
En cuanto al hallazgo clínico ocular más frecuente en estos pacientes fue el compromiso del disco óptico, a diferencia de la población adulta donde los hallazgos más frecuentes son el desprendimiento de retina e iridociclitis(18). El mecanismo fisiopatológico del mayor compromiso de disco óptico en niños no está claro; sin embargo, la identificación de este signo en la población pediátrica observada en este estudio puede orientar a oftalmólogos y especialistas en uveítis al momento de evaluar niños con sospecha de VKH e inicio oportuno de tratamiento.
En el mayor porcentaje de pacientes, la AV al ingreso fue menor a 20/200; sin embargo, posterior al tratamiento sólo 2 ojos permanecieron con una AV en rango de ceguera legal, siendo menor a lo reportado en estudio de VKH en población pediátrica alcanzando alrededor del 20%(19). Esto puede ser explicado por el advenimiento y uso de nuevos fármacos con mayor potencia inmunosupresora que controla la enfermedad. El pronóstico visual es variable, pero en general es bueno si el diagnóstico es temprano y se inicia tratamiento en forma precoz, tal como se ve en nuestros pacientes donde alrededor de un 60% logra AV final ≥ 0.6, similar a lo descrito en otras poblaciones de VKH en edades pediátricas (19).
Llama la atención el alto porcentaje de casos incompletos o presuntos, siendo más del 70% de nuestros pacientes, similar a lo descrito en población general con VKH(9). Lo anterior se podría explicar en parte por la naturaleza evolutiva de algunos de los criterios diagnósticos que comprenden manifestaciones extraoculares, las cuales pueden presentarse por primera vez en el curso de la enfermedad ya antes diagnosticada(15).
La inflamación intraocular crónica recurrente que desarrollan ciertos pacientes usualmente tras 6 a 9 meses luego de la presentación inicial, caracterizada por exacerbaciones de la uveítis granulomatosa anterior resistente al tratamiento esteroideo, da lugar al desarrollo de complicaciones: catarata subcapsular posterior, sinequias posteriores, glaucoma, edema macular, neovascularización coroidea y papilar, membrana neovascular subretiniana y ptisis bulbar(9). Las complicaciones oftalmológicas se han reportado en menos de la mitad de los pacientes con VKH(9), contrastándose con nuestros resultados que evidencian la presencia de complicaciones en 7 de cada 10 pacientes. Lo anterior reafirma la mayor agresividad de la enfermedad en niños previamente descrita, sobre todo considerando el potencial desarrollo de ceguera y ambliopía subsecuente. En cuanto a las complicaciones observadas la más frecuente fue glaucoma, mostrando una incidencia mayor a la descrita en población con VKH, la cual fluctúa alrededor del 30%(9); sin embargo, en un estudio de VKH en población pediátrica encontraron un 40% en una muestra de 20 ojos(19), por lo que da la impresión que el glaucoma es una complicación frecuente en niños con VKH. El resultado del aumento de la presión intraocular es multifactorial. Dentro de las posibles explicaciones para este fenómeno se encuentran el cierre angular secundario a la presencia de sinequias posteriores y periféricas, la respuesta inmune que genera daño en el trabéculo y glaucoma secundario al uso de esteroides(19). Por otro lado, la presencia de cataratas en nuestros pacientes resultó ser menor a la reportada en otros pacientes con VKH tanto pediátricos como adultos, donde ha resultado ser la complicación más frecuente, la cual fluctúa entre 45-61%(9,19). La etiopatogenia de la catarata es compleja y no ha sido del todo aclarada. Se han reconocido distintas mutaciones genéticas que interfieren con la síntesis de proteínas de membrana, del citoesqueleto, del cristalino, factores de transcripción y proteínas relacionas al metabolismo(20). No obstante, estos factores no han sido estudiados en pacientes con VKH, por lo que el mecanismo etiopatogénico de la catarata en esta enfermedad se desconoce. En pacientes con uveítis se plantea que la inflamación intraocular persistente, el tratamiento, particularmente el uso de corticoesteroides sistémicos y locales, y las complicaciones quirúrgicas asociadas a la uveítis serían factores que inciden en la progresión de catarata, incluso desde edades tempranas(21).
El principio del tratamiento es la supresión de la inflamación intraocular. Para esto se utiliza corticoesteroides sistémicos como primera línea, asociado o no a inmunosupresores. Las dosis, la duración del tratamiento y la pauta debe individualizarse en cada paciente(22) no exentos de complicaciones: hipertensión ocular, catarata y retardo del crecimiento(19). Debido a esto, muchos especialistas recomiendan el uso de agentes ahorradores de corticoesteroides como metotrexato y 186 Revista Hospital Clínico Universidad de Chile ciclosporina para el control de la enfermedad(13). Un alto porcentaje de nuestros pacientes requirió asociación con inmunosupresores para el control de la enfermedad. A su vez, el número de recidivas objetivadas en estos pacientes pediátricos fue mayor a lo reportado en pacientes con VKH de todas las edades, destacando además la presencia de compromiso posterior, la cual requiere un alto grado de sospecha(9). Lo anterior también apoya la mayor agresividad de la enfermedad ya advertida en población pediátrica.
El VKH es una enfermedad inflamatoria crónica con variadas manifestaciones, la cual requiere un alto grado de sospecha por parte del médico general y del especialista para la instalación de un tratamiento precoz, agresivo y prolongado. Se requiere de un equipo multidisciplinario, no sólo por el compromiso multisistémico propio de la enfermedad, sino también por el grado de inmunosupresión que se requiere para lograr su remisión.
AGRADECIMIENTOS
Los autores están profundamente agradecidos a los pacientes y los controles por su participación en este estudio. Víctor Velásquez y Pablo Romero tuvieron pleno acceso a todos los datos del estudio y se responsabilizaron de la integridad de los datos y de la precisión del análisis de los datos. Esta investigación no fue apoyada por ninguna subvención.
REFERENCIAS
1. Moorthy R S, Inomata H, Rao N A. Vogt- Koyanagi-Harada syndrome. Surv Ophthalmol 1995;39:265–92.
2. Riveros A, Romera P, Holgado S, Anglada J, Martinez M, Tejera B. Enfermedad de Vogt-Koyanagi-Harada. Semin Fund Esp Reumatol 2012;13:142–6.
3. Tabbara KF, Chavis PS, Freemann WR. Vogt-Koyanagi-Harada syndrome in children compared to adults. Acta Ophthalmol Scand 1998;76:723–6.
4. Rao NA. Mechanisms of inflammatory response in sympathetic ophthalmia and VKH syndrome. Eye 1997;11:213–6.
5. Rathinam SR, Namperumalsamy P, Nozik RA, Cunningham ET Jr. Vogt- Koyanagi- Harada syndrome after cutaneus injury. Ophthalmology 1999;106:635–8.
6. Norose K, Yano A, Wang X, Tokushima T, Umihira J, Seki A et al. Dominance of activated T cells and interleukin-6 in aqueous humor in VKH syndrome. Invest Ophthalmol Vis Sci 1994;35:33-9.
7. Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE et al. Complications and prognostic factors in Vogt- Koyanagi-Harada disease. Am J Ophthalmol 2001;131:599–606.
8. Martínez A, Eguíluz S, Artaraz J, Fonollosa A. Enfermedad de Vogt-Koyanagi-Harada: descripción de diez casos. Internistas 2013;0:17- 24.
9. Datoo O’Keefe G, Rao N. Mayor review Vogt-Koyanagi-Harada disease. Survey of Ophthalmology 2017;62:I-25.
10. Biswas J, Narain S, Das D, Ganesh SK. Pattern of uveitis in a referral uveitis clinic in India. Int Ophthalmol 1996;20:223.
11. Martin TD, Rathinam SR, Cunningham ET. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt- Koyanagi-Harada disease in South India. Retina 2010;30:1113e21.
12. Hamade IH, Shamsi Al HN, Dhibi Al H, et al. Uveitis survey in children. Br J Ophthalmol 2009;93:569.
13. Cunningham jr ET. Uveitis in children. Ocul Immunol Inflam 2000;8:251–61.
14. Rathinam SR, Vijayalakshmi P, Namperumalsamy P, Nozik RA, Cuningham ET. Vogt- Koyanagi-Harada syndrome in children. Ocul Immunol Inflamm 1998;6:155-61.
15. Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S et al. Revised Diagnostic Criteria for Vogt Koyanagi Harada Disease: Report of an International Committee on Nomenclature. Am J Ophthalmol 2001;131:647-52.
16. Tsai JH, Evans M, Rao NA. Comparative study of two sets of criteria for the diagnosis of Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2006;141:778–9.
17. World Health Organisation. International Statistical Classification of Diseases and Related Health Problems. 2011:53–4.
18. Giordano V, Schlaen A, Guzman M, Couto C. Spectrum and visual outcomes of Vogt- Koyanagi-Harada disease in Argentina. Int J Ophlthalmol 2017;10:98-102.
Correspondencia:
Dr. Pablo Romero Carrasco
Departamento de Oftalmología, Hospital Clínico Universidad de Chile
[email protected]
562 2978 8539 / 562 2978 9427