Abstracts Publicaciones 2021
Quiénes somos
Instrucciones a autores
Responsabilidad autoría
Contacto
Portal Revistas U. de Chile
Nuevas estrategias farmacológicas en la enfermedad de Alzheimer: la hiperactividad y la neuroinflamación como nuevos blancos potenciales
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the progressive deterioration of cognitive functions such as memory, language, executive function, and visuospatial function, starting from the late ages of life (older than 65 years). The etiology of AD is multifactorial involving the interaction of genetic, environmental, and systemic factors. Histopathologically characterized by the progressive accumulation of peptide Aβ and TAU protein. Markers of damage that begin to accumulate decades before symptoms can be detected. However, during the early stages of the disease, systemic inflammation and cortical/hippocampal hyperactivity have been revealed present in patients. In the following review, we present a perspective on how neuroinflammatory processes and pathological cortical hyperexcitability contribute to the onset and development of the early stages of AD. We present a current summary of the basic and clinical evidence that allows us to understand the complexity and heterogeneity of this pathology. Finally, we briefly collect and analyze the main therapeutic strategies that have been studied to reduce the phenotype of neuronal hyperactivity directly or indirectly.
Rev Hosp Clín Univ Chile 2023; 34: 109 - 22 / DOI: 10.5354/2735-7996.2023.71414
Pedro Lobos Z., Jamileth More C., Bárbara Bruna J., Antonello Penna S.
Enfermedad de Alzheimer: diagnóstico clínico y biomarcadores
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa progresiva que se manifiesta por graves alteraciones a nivel cognitivo y de la memoria hasta alcanzar la completa pérdida de la independencia y finalmente, la muerte(1,2). Se estima que para el año 2050 sólo esta enfermedad pueda afectar a más de 50 millones de personas a nivel global(3,4). Actualmente los costos económicos asociados al tratamiento y pérdida de productividad debido a la EA y otras demencias alcanzan los US$ 321 MM(5). Para los países de Latinoamérica, incluyendo Chile, este problema es aún más grave. Estudios recientes estiman que la prevalencia de demencias entre las personas mayores de 65 años oscila entre el 7,1% y 11,5%, cifras que probablemente están drásticamente subestimadas debido a la falta de presupuesto, infraestructura y personal capacitado para el diagnóstico apropiado de la enfermedad y que podrían al menos triplicarse para el año 2050(6,7).
Múltiples factores ambientales y biológicos están vinculados al desarrollo de la EA; sin embargo, la patología subyacente aún no se ha entendido por completo. La gran mayoría de las personas que desarrollan EA tienen 65 años o más y sufren la denominada EA de inicio tardío. Por otro lado, otras personas comienzan con demencia clínica a edades más tempranas, padeciendo la denominada EA familiar o de herencia dominante, caracterizada por involucrar mutaciones en los genes de la proteína precursora amiloide (APP, por sus siglas en inglés) y de las proteínas presenilinas 1 y 2 (PSEN1 y PSEN1)(1,8). Hoy en día sabemos que la EA puede comenzar décadas antes del diagnóstico clínico, presentándose como un detrimento en las capacidades cognitivas, insuficiente para constituir una demencia debido a la EA, pero mayor que lo esperado en el envejecimiento normal. Esta etapa es denominada deterioro cognitivo leve (DCL)(9).
El diagnóstico de DCL y demencia es principalmente clínico, basado en la historia y exámenes de laboratorio que permitan descartar factores confundentes. Las herramientas diagnósticas clínicas más utilizadas son el Mini-Mental State Examination (MMSE), el Montreal Cognitive Assessment test (MoCA test) y la Escala Clínica de Demencia - Suma de Cajas (CDR-SB, considerada el estándar de oro). Estas herramientas ponderan distintas funciones psicométricas relacionadas a la memoria, atención, cálculo, orientación, entre otros factores, otorgándoles un puntaje que luego discrimina entre DCL, demencia leve y demencia tipo Alzheimer(10–12). El diagnóstico clínico se puede complementar con el uso de biomarcadores para la EA, como son la medición de los niveles en líquido cefalorraquídeo del péptido amiloide y proteína TAU total o fosforilada y/o la detección de amiloide en el cerebro mediante tomografía de emisión de positrones (PET, por sus siglas en inglés). Estas técnicas son más invasivas y conllevan un alto costo, razón por la cual en la actualidad se han hecho crecientes esfuerzos en la búsqueda de posibles nuevos biomarcadores especialmente para etapas tempranas de la EA(13).
En la presente revisión abordaremos la evidencia preclínica y clínica que permite conectar los mecanismos asociados a la fisiopatología temprana de la EA con la hiperexcitabilidad neuronal y la neuroinflamación. Además, mencionamos y analizamos brevemente las principales estrategias terapéuticas que se han estudiado para reducir directa o indirectamente el fenotipo de hiperactividad neuronal (Figura 1).
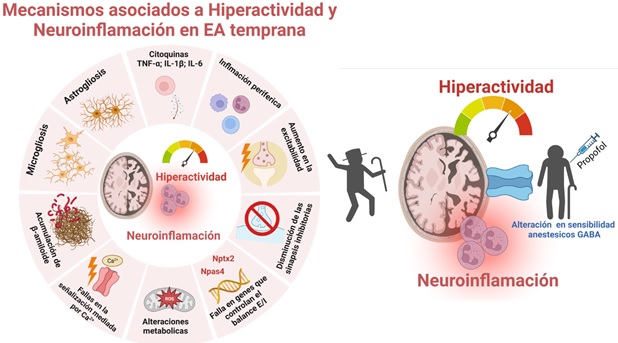
Figura 1. Resumen de la evidencia del rol de hiperactividad y neuroinflamación en la EA y resumen del estudio clínico propuesto. A la izquierda se esquematizan algunos de los mecanismos asociados al fenotipo de hiperactividad y aumento de la neuroinflamación citados en el texto. Muchos de los mediadores de estos procesos se encuentran en estudio como blancos farmacológicos o biomarcadores tempranos de la EA (ver Tablas 1 y 2). A la derecha, se propone como mecanismo de la alteración a la sensibilidad al anestésico general propofol los procesos de hiperactividad y neuroinflamación que se generan en las etapas iniciales de la EA. Esquema creado con BioRender.com.
Bases y teorías biológicas de la fisiopatología de la enfermedad de Alzheimer
La etiopatogenia de la EA más aceptada hasta el momento corresponde a la denominada hipótesis de la “cascada amiloide”, la cual establece que la acumulación y agregación progresiva del péptido Aβ desencadena la muerte neuronal en las zonas del cerebro degeneradas en la EA(14). El péptido Aβ se genera por un procesamiento anormal de la APP, mediado por los complejos enzimáticos β-secretasa y γ-secretasa. Los monómeros Aβ en medio acuoso rápidamente dan lugar a varios tipos de agregados, que incluyen los oligómeros solubles de Aβ (AβOs), protofibrillas y fibrillas amiloideas de alto peso molecular, siendo estas últimas las que se depositan formando las placas seniles(15,16). En las últimas décadas, se ha determinado que los AβOs, por sobre las fibrillas y agregados de alto peso molecular, son las principales neurotoxinas que están presentes desde etapas tempranas de la enfermedad, acumulándose en conglomerados mayores que interrumpen progresivamente distintas funciones neuronales(17–19).
La preponderancia patológica, a lo menos inicial, del péptido Aβ por sobre la proteína TAU se ha corroborado en diferentes modelos tanto in vitro como in vivo. La acumulación del péptido Aβ precede y desencadena la propagación de la patología TAU(20–22). Además, se ha determinado que la patología Aβ y TAU son sinérgicas entre sí, desencadenando distintos procesos patológicos claves en distintas etapas de la EA. La visión actual propone que inicialmente se desarrolla una hiperactividad cerebral en la EA temprana debido principalmente a la patología Aβ. Luego, la EA progresa a una marcada hipoactividad debido a la patología combinada Aβ más TAU, convergiendo particularmente en vías de señalización críticas a nivel de cambios en la expresión génica que terminan por potenciar varios mecanismos fundamentales tempranos para el desarrollo de la EA(23).
Análisis de los resultados de los últimos fármacos aprobados y necesidad de la búsqueda de nuevos biomarcadores enfocados en la patología temprana de la EA.
Actualmente existen al menos 143 agentes en investigación en fases clínicas, como posibles tratamientos contra la EA (31 en fase III, datos enero 2022)(24); sin embargo, actualmente solo existen siete medicamentos aprobados por la FDA para tratar la EA. Cinco de los cuales —donepezilo, rivastigmina, galantamina, memantina y memantina combinada con donepezilo— sólo ayudan temporalmente con los síntomas de la enfermedad, pero no intervienen en la biología subyacente ni alteran el curso de la enfermedad(25). Los últimos dos fármacos en salir al mercado —mediante el registro de aprobación condicional acelerada— corresponden a los anticuerpos monoclonales dirigidos contra conglomerados de Aβ. Estos son lecanemab (Lequembi® aprobado enero 2023) y aducanumab (Adulhem® aprobado junio 2021). Ambos fármacos corresponden a las primeras terapias aprobadas que se dirigen contra la biología subyacente de la EA; no obstante, la aprobación acelerada se justificó principalmente por el potente efecto reductor del Aβ cerebral en desmedro de la eficacia clínica(26–28).
En particular para el caso de aducanumab, su evidencia se centra en los resultados de los ensayos clínicos ENGAGE y EMERGE, los cuales sólo demostraron retrasar levemente el avance del deterioro cognitivo de los pacientes con EA inicial y que en conjunto a su elevado precio en el mercado, provocó serias dudas acerca de su costo-beneficio(27,29). El caso de lecanemab fue bastante similar, para el cual el ensayo clínico CLARITY logró demostrar una mayor disminución de Aβ y un efecto algo mayor en el retraso del deterioro cognitivo. El cambio medio del CDR-SB entre los grupos ajustados desde el inicio hasta los 18 meses fue de +1,21 con lecanemab y de +1,66, con placebo (diferencia −0,45 (intervalo de confianza del 95 % −0,67 a −0,23), P <0,001). No obstante, no queda claro si este retraso en el progreso de la enfermedad es clínicamente significativo(28,30). En conjunto, el análisis de todos los ensayos con los principales anticuerpos anti-amiloide indican una relación entre el nivel de reducción de Aβ (determinación por PET) y los beneficios clínicos evaluados mediante la escala CDR-SB, lo que sugiere que efectivamente la reducción de Aβ puede en alguna medida lograr alterar el curso de la enfermedad(31). Este es un avance promisorio que no ha sido alcanzado, al menos por el momento, por ninguna otra terapia en desarrollo y renueva las esperanzas en hipótesis de la cascada amiloide. Es posible especular que la diferencia en la eficacia lograda entre ambos fármacos, en favor de lecanemab, puede deberse a la mayor selectividad de lecanemab en la unión contra las conformaciones de Aβ más tóxicas (solubles y de mayor tamaño) que se producen en las etapas más tempranas de la EA y, por lo tanto, reduce la formación de placas de Aβ como la neurotoxicidad de las formas solubles(32,33).
Sin embargo, como toda terapia, no está exenta de efectos adversos, siendo el más importante la denominada anomalías de imagen relacionadas con amiloide (ARIA, por sus siglas en inglés). Las ARIA representan sitios relacionados con la fuga de contenido intravascular, generando edema o microhemorragias cerebrales relacionados a los cambios en el contenido de amiloide cerebral(34). Tanto lecanemab como aducanumab presentan el riesgo de generar la aparición de ARIA, aunque la incidencia notificada de ARIA en los ensayos de lecanemab es menor en comparación con aducanumab(35). No obstante, el informe reciente del caso fatal de un paciente que sufrió un accidente cerebrovascular isquémico relacionado a una hemorragia intracerebral y vasculitis, debido al tratamiento conjunto de lecanemab y trombólisis intravenosa, puso en duda la seguridad de estas terapias y resalta aún más la importancia de considerar factores tanto individuales como la administración concomitante de otros fármacos que pudiesen promover estos efectos graves(36,37).
Lecanemeab y aducanumab representan un ejemplo más de las dificultades que aparecen al intentar trasladar los avances de la ciencia básica-preclínica en la obtención de terapias y/o biomarcadores para la EA. Entre las limitaciones que es habitual encontrar y que dificultan enormemente el éxito de estos ensayos, destacan: un fenotipo y población altamente heterogénea, inicio del tratamiento en etapas tardías, duración extensa (al menos 1-2 años de seguimiento), detención prematura ensayos clínicos, entre otros(38).
Además, resulta clave la selección adecuada de los pacientes, de tal manera de disminuir la alta variabilidad fenotípica, por lo cual es necesario descubrir y desarrollar urgentemente nuevos biomarcadores para la EA que pueden revelar el grado de disfunción sináptica característico de las etapas tempranas de esta enfermedad. Actualmente, el diagnóstico de la EA es principalmente clínico, basado en cuestionarios estandarizados; mientras que las pruebas de laboratorio apoyan al diagnóstico, pero son invasivas y/o de alto costo. En este sentido, los biomarcadores plasmáticos son prometedores dado que son de más fácil realización.
A la fecha, los biomarcadores plasmáticos más promisorios son los fragmentos solubles o epítopos específicos de proteínas como Tau181 fosforilado (P-tau181), neurofilamento (NFL), la razón de amiloide-β (Aβ42/40) y la proteína ácida fibrilar glial (GFAP). Dada la escasa concentración en que están presentes en las muestras utilizadas en la clínica, es altamente recomendable detectarlos utilizando tecnologías con la sensibilidad y exactitud apropiadas que permitan optimizar las valiosas muestras obtenidas. Un ejemplo cada vez más utilizado en la literatura es la tecnología de detección de matriz de molécula única (Simoa®), que analizando combinaciones de biomarcadores ha logrado discriminar significativamente entre los estados de DCL, EA y demencia(39,40). Es importante señalar que recientemente esta tecnología está disponible para la realización de estudios clínicos en el Hospital Clínico Universidad de Chile.
Hiperactividad en circuitos córtico-hipocampales: un marcador temprano del desarrollo de la EA (básico-clínico)
Múltiples líneas de evidencia, incluyendo desde modelos de roedores hasta humanos, han proporcionado un fuerte apoyo a la idea de la que la EA se inicia tempranamente con la aparición de la interrupción patológica de los circuitos córtico-hipocampales. En particular, se ha determinado un fenotipo de hiperactividad neuronal, producto de un estado de hiperexcitabilidad intrínseca neuronal acentuado, en las etapas tempranas presintomáticas de pacientes con EA familiar(41). A nivel preclínico, se ha establecido una estrecha relación entre la producción del péptido Aβ y el nivel de actividad neuronal, donde la hiperactividad neuronal induce la producción de más péptido Aβ, lo que provoca una mayor excitabilidad, produciendo un ciclo vicioso de potenciación mutua(42). No obstante, se ha visto un rol disímil entre las especies de Aβ y TAU, donde la acumulación de especies de Aβ que preceden a la aparición de TAU inducen un fenotipo de hiperactividad en circuitos córtico-hipocampales en las etapas tempranas de la EA. Así, la acumulación más tardía de TAU se relaciona con un fenotipo de hipoactividad más prevalente en etapas tardías de la EA(43).
Entre los marcadores más incuestionables que demuestren la hiperactividad temprana en la EA encontramos la actividad epileptogénica, la cual se ha observado que puede surgir varios años antes de los síntomas y/o el deterioro cognitivo y persiste en la fase de demencia(44,45). En la práctica clínica, las convulsiones en pacientes con EA pueden pasar desapercibidas fácilmente porque generalmente se presentan como convulsiones sin actividad motora y pueden superponerse con otros síntomas de la enfermedad. La presencia de actividad epiléptica subclínica, registrada con un electroencefalograma (EEG) se ha relacionado con una aceleración del deterioro cognitivo en pacientes con EA(46,47). Esto se ha corroborado recientemente en un metaanálisis, en el cual se detectó una correlación positiva bidireccional entre la prevalencia de epilepsia y la EA(48).
Anormalidades en la señalización inhibitoria GABAérgica durante la EA: Observaciones clínicas y preclínicas
Varias líneas de investigación apuntan a una falla en el tono de excitación/inhibición, particularmente en las sinapsis inhibitorias de interneuronas GABAésrgica, como un evento temprano en la patología de la EA(49). En particular, se ha observado que la disminución de la duración y calidad del sueño en adultos se relaciona con aumento en la aparición de demencia(50), determinándose que la reducción del sueño promueve la acumulación del péptido Aβ por interrupción de los mecanismos de aclaramiento linfático(51). De hecho, un trabajo reciente determinó que los oligómeros de Aβ reducen el sueño NREM (non-rapid eye movement) y disminuyen la respuesta a anestésicos generales agonistas de los receptores GABA(A), como el isoflurano o propofol en modelos transgénicos de la EA(52). Existen varios reportes que corroboran una respuesta disminuida a los anestésicos generales en modelos de ratones transgénicos de la EA(53–56). En suma, la actividad inhibitoria GABAérgica estaría disminuida en etapas tempranas de la EA.
Se ha observado fallas en la señalización por calcio y segundos mensajeros en neuronas y células gliales(57,58), lo que es importante para el control adecuado de la plasticidad homeostática controlada por factores como Npas4 y Nptx2, los que controlan las sinapsis inhibitorias y excitatorias(59).
Los resultados del grupo de Penna y cols. han mostrado que los pacientes adultos mayores con deterioro cognitivo que se someten a una cirugía bajo una anestesia general con agentes GABAérgicos tienen una menor actividad en el EEG frontal cuando se encuentran bajo los efectos de los anestésicos generales GABAérgicos, tales como sevofluorano y propofol(60–62). El seguimiento de la actividad por EEG frontal se ha realizado, usando monitores utilizados para guiar la dosificación de la anestesia en la clínica. El más ampliamente utilizado y que tiene un mayor respaldo en las guías y recomendaciones clínicas es el monitor BIS® Covidien (Medtronic, Minneapolis). Este monitor entrega un índice propietario derivado del procesamiento de la señal del EEG frontal, denominado índice biespectral (BIS, por sus siglas en inglés), y además adquiere la señal cruda del EEG frontal en su conjunto, otorgando la información para establecer adecuadamente el nivel de actividad cortical. Esto ha permitido plantear la hipótesis de que los pacientes con deterioro cognitivo son más sensibles al efecto de los anestésicos generales GABAérgicos; sin embargo, los mecanismos celulares y moleculares que intervienen en esta relación aún se encuentran en estudio.
Se están ejecutando ensayos clínicos con el objetivo de probar la eficacia y seguridad de estrategias para reducir la sobreexcitación neuronal y mejorar la cognición. En estos ensayos se están utilizando fármacos antiepilépticos como levetiracetam, estimulación sensorial para potenciar la actividad en la frecuencia gamma o inmunoterapia contra especies de AβOs que impiden la hiperactividad (Tabla 1). Los resultados publicados recientemente(63) para el uso de levetiracetam, si bien no mostraron una mejora significativa en la función cognitiva en pacientes con EA, los análisis posteriores mostraron que el tratamiento sí mejoró la función ejecutiva y la memoria espacial en el subgrupo de los participantes de EA con actividad epileptiforme(63). Actualmente, estos resultados están siendo validados en una población de estudio más grande (Clinical Trial: NCT03875638). Por último, un estudio reciente(64) demostró que los pacientes con EA y actividad epileptiforme presentaron una actividad oscilatoria reducida en la actividad del EEG a nivel de la banda alfa (8-12 Hz) y aumentada en la banda delta-theta (2-8 Hz), en comparación con pacientes con EA y sin actividad epileptiforme, lo que muestra que se pueden identificar a los pacientes con EA y actividad epileptiforme(65).

En conclusión, lo observado en modelos murinos y en humanos nuevamente no es concordante. Los modelos animales de EA serían más resistentes al efecto de los anestésicos generales, mientras que los humanos con deterioro cognitivo son más sensibles. Probablemente, la explicación se debe a que los modelos animales tienen un nivel de hiperexcitabilidad aumentada, mientras que los humanos no tienen dicha alteración en la excitabilidad. Para demostrar esto, se deben realizar nuevos estudios tanto clínicos como preclínicos.
La neuroinflamación y su contribución al fenómeno de hiperactividad en la EA
Otro proceso estrechamente asociado con el progreso desde etapas tempranas de la EA y otras enfermedades neurodegenerativas es la neuroinflamación. Estudios clínicos han evidenciado niveles elevados de citoquinas proinflamatorias (IL-1β, IL-1, IL-6, TNF-α), tanto en suero como en tejidos aislados de cerebros post mortem de pacientes con EA(66,67). Los diversos trabajos han mostrado que las citoquinas proinflamatorias como TNF-α promueven la hiperactividad y excitotoxicidad por distintos mecanismos(68). De hecho, se ha determinado que estas citoquinas pueden actuar inhibiendo específicamente el tono inhibitorio GABAérgico y, por lo tanto, promoviendo indirectamente mayor hiperactividad y la patología temprana de Alzheimer(49). Especialmente de interés son los estudios recientes que han mostrado que distintos procesos que producen neuroinflamación inducen alteraciones en la actividad neuronal. Específicamente, la interrupción patológica de la vía mTOR en la microglía se relacionó con la producción de descargas epilépticas(69) y se ha comprobado que la activación de las microglías inhiben la actividad neuronal, mediante la captación de ATP/ADP producto del metabolismo neuronal, conectando la actividad metabólica con la neuroinflamación(70). Por otro lado, se determinó que la expresión del receptor metabotrópico GABA(B) en las microglías durante diversas etapas del desarrollo de las neuronas corticales potencian las sinapsis inhibitorias(71).
En su conjunto, estos antecedentes parecen mostrar que la neuroinflamación crónica, mediante el aumento sostenido de citoquinas proinflamatorias y la activación patológica de las microglías(72), tiende a desarrollar un aumento de la actividad eléctrica cerebral por mecanismos que pueden involucrar la interrupción del tono inhibitorio GABAérgico y/o un aumento en el tono excitatorio glutamatérgico. Esto promueve un fenotipo de hiperexcitabilidad neuronal y glial patológico que puede promover la cascada patológica inicial de la EA(41,58,73). Por este motivo, no es de sorprender que muchos de los principales nuevos blancos farmacológicos se dirigen a propiedades y funciones microgliales específicas, tales como actividad inflamatoria, fagocitosis, proliferación, metabolismo o vigilancia(74). En la Tabla 2, se resumen estudios preclínicos que han estudiado mecanismos para disminuir la hiperactividad.

CONCLUSIONES
Para lograr un mejor entendimiento de la fisiopatología de la EA, debemos considerar una visión más actual e integrativa acerca de los procesos patológicos que suceden en las etapas tempranas de la EA, alejándonos de la clásica perspectiva lineal para lograr acercarnos a la complejidad que surge de la interacción entre la patología Aβ y TAU, entendiendo que convergen en múltiples mecanismos que se potencian mutuamente entre sí(23). Los estudios clínicos de la inmunoterapia anti-amiloide, aunque lejos de ser exitosos, parecen lograr enlentecer la progresión de la enfermedad y, por lo tanto, la reducción de la carga de péptido Aβ puede ser la primera estrategia que en alguna medida logre alterar el curso de la enfermedad. No obstante, para mejorar la efectividad y seguridad de estas terapias es necesario estudiar nuevos mecanismos que se alejen de los paradigmas actuales. En particular se ha expandido la mirada desde una centrada en las células neuronales a una en que contribuirían simultáneamente diferentes tipos celulares, incluyendo la activación aberrante de células gliales y la inclusión decisiva de mecanismos con el aumento de la neuroinflamación(58,72) y la hiperexcitabilidad en etapas tempranas de la EA(41). Entender los mecanismos que intervienen y terminan por desbordar el control de la actividad neuronal puede tener profundas implicaciones para resolver la heterogeneidad de esta patología y para el desarrollo de terapias y diagnósticos personalizados para su tratamiento(73,75).
REFERENCIAS
1. Cummings JL. Alzheimer’s disease. New England Journal of Medicine 2004; 351:56- 67.
2. David S Knopman, Helene Amieva, Ronald C Petersen, Gäel Chételat, David M Holtzman, Bradley T Hyman et al. Alzheimer disease. Nat Rev Dis Primers 2001;7:33.
3. GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022;7:e105–e125.
4. 2022 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2022;18: 700–89.
5. Kinchin I, Mitchell E, Agar M, Trépel D. The economic cost of delirium: A systematic review and quality assessment. Alzheimer’s and Dementia 2021;17:1026–41.
6. Manes F. The huge burden of dementia in Latin America. The Lancet Neurology 2016;15:29.
7. Mario Alfredo Parra, Sandra Baez, Lucas Sedeño, Cecilia Gonzalez Campo, Hernando Santamaría-García, Ivan Aprahamian et al. Dementia in Latin America: Paving the way toward a regional action plan. Alzheimer’s & Dementia 2021;17:295–313.
8. Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006296.
9. Marilyn S. Albert, Steven T. DeKosky, Dennis Dickson, Bruno Dubois, Howard H. Feldman, Nick C. Fox et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:270–9.
10. Folstein MF, Folstein SE, McHugh PR. Mini-mental state. J Psychiatric Research 1975;12:189–98.
11. Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 1993;43:2412.2-2412-a.
12. Delgado C, Araneda A, Behrens MI. Validation of the Spanish-language version of the Montreal Cognitive Assessment test in adults older than 60 years. Neurologia 2019;34:376–85.
13. Zetterberg H, Schott JM. Biomarkers for Alzheimer’s disease beyond amyloid and tau. Nature Medicine 2019;25:201–3.
14. Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991;6:487–98.
15. Glabe CG. Structural classification of toxic amyloid oligomers. J Biological Chemistry 2008;283:29639–43.
16. Ahmed M, Davis J, Aucoin D, Sato T, Ahuja
S, Aimoto S et al. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nature Structural & Molecular Biology 2010;17:561–7.
17. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nature Reviews Molecular Cell Biology 2007;8:101–12.
18. Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer`s disease. Frontiers in Cellular Neuroscience 2015;9:1–17.
19. Habashi M, Vutla S, Tripathi K, Rahimipour S. Early diagnosis and treatment of Alzheimer’s disease by targeting toxic soluble Aβ oligomers. Proc Natl Acad Sci USA 2022;119:e2210766119.
20. Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E et al. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO Journal 2013;32:2920–37.
21. Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke, SE et al. Alzheimer’s disease-like pathology induced by amyloid- oligomers in nonhuman primates. J Neuroscience 2014;34:13629–43.
22. Jana MK, Cappai R, Pham CLL, Ciccotosto GD. Membrane-bound tetramer and trimer Abeta oligomeric species correlate with toxicity towards cultured neurons. J Neurochemistry 2016;136:594–608.
23. Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nature Neuroscience 2020;23:1183–93.
24. Cummings J, Lee G, Nahed P, Nojoo Kambar MEZ, Zhong K, Fonseca J et al. Alzheimer’s disease drug development pipeline: 2022. A&D Transl Res & Clin Interv 2022;8.
25. Birks J. Cholinesterase inhibitors for Alzheimer’s disease (Review). The Cochrane Library 2012;1–51.
26. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50–6.
27. Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s Disease. J Prev Alz Dis 2022;9:197–210.
28. Van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M et al. Lecanemab in early Alzheimer’s disease. N Engl J Med 2023;388:9–21.
29. Mahase E. FDA considers Alzheimer’s drug previously abandoned for lack of effectiveness. BMJ 2021;367:6164.
30. Mahase E. Lecanemab trial finds slight slowing of cognitive decline, but clinical benefits are uncertain. BMJ 2022;379:o2912.
31. Perneczky R Jessen F, Grimmer T, Levin J, Flöel A, Peters O et al. Anti-amyloid antibody therapies in Alzheimer’s disease. Brain 2023;146:842-9.
32. Söderberg, L. et al. Lecanemab, aducanumab, and gantenerumab — binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics 2023;20:195–206.
33. Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE 2012;7:e32014.
34. Barakos J, Sperling R, Salloway S, Jack C, Gass A, Fiebach JB et al. MR imaging features of amyloid-related imaging abnormalities. Am J Neuroradiol 2013;34:1958–65.
35. Smith EE, Greenberg SM, Black SE. The impending era of beta-amyloid therapy: Clinical and research considerations for treating vascular contributions to neurodegeneration. Cerebral Circulation - Cognition and Behavior 2023;100159.
36. Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 2022;79:13.
37. Reish NJ, Jamshidi P, Stamm B, Flanagan ME, Sugg E, Tang M et al. Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t-PA for stroke. N Engl J Med 2023;388:478–9.
38. Haass C, Selkoe D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol 2022;20:3001694.
39. Simrén J, Leuzy A, Karikari TK, Hye A, Benedet AL, Lantero-Rodriguez J et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimer’s & Dementia 2021;17:1145–56.
40. Chatterjee P, Pedrini S, Doecke JD, Thota R, Villemagne VL, Doré V et al. Plasma Aβ42/40 ratio, p-tau181, GFAP, and NfL across the Alzheimer’s disease continuum: A cross-sectional and longitudinal study in the AIBL cohort. Alzheimer’s & Dementia. Dement 2023;19:1117–34.
41. Targa Dias Anastacio, Matosin N, Ooi L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry 2022;12:257.
42. Zott B, Simon MM, Hong W, Unger F, Chen-Engerer H, Frosch MP et al. A vicious cycle of β amyloid−dependent neuronal hyperactivation. Science 2019;365:559–65.
43. Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci 2019;22:57–64.
44. Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. The Lancet Neurology 2017;16:311– 22.
45. Giorgi FS, Saccaro LF, Busceti CL, Biagioni F, Fornai F. Epilepsy and Alzheimer’s Disease: Potential mechanisms for an association. Brain Research Bulletin 2020;160:107–20.
46. Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease: Subclinical Epileptiform Activity in AD. Ann Neurol 2016;80:858–70. 124 Revista Hospital Clínico Universidad de Chile
47. Horvath AA, Papp A, Zsuffa J, Szucs A, Luckl J, Radai F et al. Subclinical epileptiform activity accelerates the progression of Alzheimer’s disease: A long-term EEG study. Clinical Neurophysiology 2021;132:1982–9.
48. Dun C, Zhang Y, Yin J, Su B, Peng X, Liu L. Bi-directional associations of epilepsy with dementia and Alzheimer’s disease: a systematic review and meta-analysis of longitudinal studies. Age and Ageing 2022;51:afac010.
49. Bi D, Wen L, Wu Z, Shen Y. GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimer’s disease. Alzheimer’s & Dementia 2020;16:1312–29.
50. Sabia S, Fayosse A, Dumurgier J, van Hees VT, Paquet C, Sommerlad et al. Association of sleep duration in middle and old age with incidence of dementia. Nat Commun 2021;12:2289.
51. Winer JR, Mander BA, Kumar S, Reed M, Baker SL, Jagust WJ et al. Sleep disturbance forecasts β-amyloid accumulation across subsequent Years. Current Biology 2020;30:4291-8 e3.
52. Zarhin D, Atsmon R, Ruggiero A, Baeloha H, Shoob S, Scharf O et al. Disrupted neural correlates of anesthesia and sleep reveal early circuit dysfunctions in Alzheimer models. Cell Reports 2022;38:110268.
53. Bianchi S L, Caltagarone BM, La Ferla FM, Eckenhoff RG, Kelz MB. Inhaled anesthetic potency in aged Alzheimer mice. Anesthesia & Analgesia 2010;110:427–30.
54. Eckel B, Richtsfeld M, Starke, L, Blobner M. Transgenic Alzheimer mice have a larger minimum alveolar anesthetic concentration of isoflurane than their nontransgenic littermates. Anesthesia & Analgesia 2010;110:438–41.
55. Seitz D, Hussain M, Eckenhoff R, Berger M. General anesthetic and the risk of dementia in elderly patients: current insights. CIA 2014;2014:1619.
56. Belrose JC, Noppens RR. Anesthesiology and cognitive impairment: a narrative review of current clinical literature. BMC Anesthesiol 2019;19:241.
57. Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Archiv European Journal of Physiology 2010;459:441–9.
58. Shah D, Gsell W, Wahis J, Luckett MS, Jamoulle T, Vermaercke B et al. Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer’s disease. Cell Reports 2022;40:111280.
59. Styr B, Slutsky I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nature Neuroscience 2018;21:463–73.
60. Gutierrez R, Egaña JI, Saez I, Reyes F, Briceño C, Venegas M et al. Intraoperative low alpha power in the electroencephalogram is associated with postoperative subsyndromal Delirium. Frontiers in Systems Neuroscience 2019;13:1–9.
61. Gutiérrez RG, Egaña JI, Maldonado FA, Sáez IA, Reyes FI, Soulat H et al. Association between lower preoperative cognition with intraoperative electroencephalographic features consistent with deep states of anesthesia in older patients: an observational cohort study. Anesthesia and Analgesia 2021;133:205–14.
62. Gutiérrez R, Maldonado F, Egaña JI, Penna, A. Electroencephalographic alpha and delta oscillation dynamics in response to increasing doses of propofol. J Neurosurgical Anesthesiology 2022;34:79–83. www.redclinica.cl 125
63. Vossel K, Ranasinghe KG, Beagle AJ, La A, Pook KA, Castro M et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurol 2021;78:1345-54. 64. Ranasinghe KG, Verma P, Cai C, Xie X, Kudo K, Gao X et al. Altered excitatory and inhibitory neuronal subpopulation parameters are distinctly associated with tau and amyloid in Alzheimer’s disease. Elife 2022;11:e77850
65. Lam AD, Shafi MM. Towards a coherent view of network hyperexcitability in Alzheimer’s disease. Brain 2022;145:423–5.
66. Chitnis T, Weiner HL. Series Editors: Marco Colonna and David Holtzmann CNS inflammation and neurodegeneration. J clinical Investigation 2017;127:1–11.
67. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2018;4:575–90.
68. Probert L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015;302:2–22.
69. Zhao XF, Liao Y, Maraj Alam M, Mathur R, Feustel P, Mazurkiewicz JE et al. Microglial mTOR is neuronal protective and anti-epileptogenic in the pilocarpine model of temporal lobe epilepsy. J Neuroscience 2020;40:7593-7608.
70. Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A et al. Negative feedback control of neuronal activity by microglia. Nature 2020;586:417–23.
71. Favuzzi E, Huang S, Saldi GA, Datta SR, Stevens B. Article GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 2021;184:4048–63.
72. Sarlus H, Heneka MT. Microglia in Alzheimer’s disease Find the latest version: Microglia in Alzheimer’s disease. J Clinical Investigation 2017;127:3240–9.
73. Harris SS, Wolf F, de Strooper B, Busche MA. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer’s Disease. Neuron 2020;107:417–35.
74. Šimončičová E, Gonçalves de Andrade E, Vecchiarelli HA, Oluleke Awogbindin I. Present and future of microglial pharmacology. Trends in Pharmacological Sciences 2022;43:669–85.
75. Hafizi S, Rajji TK. Modifiable risk factors of dementia linked to excitation-inhibition imbalance. Ageing Research Reviews 2023;83:101804.
76. Yao J, Liu Y, Sun B, Zhan X, Estillore JP, Turner RW et al. Increased RyR2 open probability induces neuronal hyperactivity and memory loss with or without Alzheimer’s disease–causing gene mutations. Alzheimer’s & Dementia 2022;18:2088–98.
77. Gourmaud S, Stewart DA, Irwin DJ, Roberts N, Barbour AJ, Eberwine G et al. The role of mTORC1 activation in seizure-induced exacerbation of Alzheimer’s disease. Brain 2022;145:324–39.
Correspondencia:
 Dr. Antonello Penna Silva
Dr. Antonello Penna Silva
 Departamento de Anestesiología y Medicina Perioperatoria, HCUCH
Departamento de Anestesiología y Medicina Perioperatoria, HCUCH
 [email protected]
[email protected]
 562 2978 8209
562 2978 8209