Abstracts Publicaciones 2021
Quiénes somos
Instrucciones a autores
Responsabilidad autoría
Contacto
Portal Revistas U. de Chile
Insomnio como síntoma precoz en enfermedad de Creutzfeldt-Jakob esporádica: presentación de 3 casos
In Chile a high incidence of Creutzfeldt – Jakob disease (CJD) has described. Insomnia is a symptom poorly studied in this disease, probably masked by the severity of other alterations. It contrasts with fatal familiar insomnia (IFF), another prionic disease with predominant involvement of thalamus, in which insomnia is a key symptom. We present three patients with clinical features and images compatible with sporadic CJD. All of them begun with severe and progressive insomnia, which preceded the final diagnosis by several months; this was associated with nocturnal psychomotor agitation and hypnagogic hallucinations. All patients died due to complications from severe dementia. Polysomnography was performed in one of the patients, which showed severe disturbance in sleep architecture with a predominance of light sleep, low sleep efficiency caused by large number of awakenings; absence of sleep spindles, vertex waves, and K complexes. In all cases caudate nucleus, thalamus, putamen and brain cortex sowed hyperintensities on T2 and diffusion weigthed brain MRI. Discussion: Insomnia was an important and early symptom in our patients with CJD. Associated with motor agitation, hallucinations, and one of them showed significant disturbance in the sleep architecture, with no spindles and vertex waves; findings also typically described in IFF. Probably the involvement of thalamic structures is a common pathophysiological pathway between these two diseases.
Rev Hosp Clin Univ Chile 2014; 25(2): 119-26
Mario Díaz S., Carlos Zúñiga V., Pablo Salinas C., Pablo González R., Ledda Aguilera O.
La enfermedad de Creutzfeldt-Jakob es una encefalopatía espongiforme transmisible, provocada por la alteración espontánea (forma esporádica) o por una mutación heredable (forma familiar) del gen que sintetiza la proteína priónica (PrP), generando un cambio en su configuración espacial (PrPsc) que le confiere la capacidad de ser patógena a través de mecanismos no del todo conocidos, dentro de los que destaca: la PrPsc es resistente a la degradación por enzimas, forma depósitos intraneuronales, induce el cambio configuracional de otras proteínas, generando muerte celular masiva en la corteza cerebral, ganglios basales y en algunos casos en el cerebelo. Además existe la posibilidad de contagio a través del contacto accidental con encéfalos de pacientes afectados.
Chile tiene una de las tasas de incidencia más altas del mundo de ECJ, alcanzando los 3 casos-año/millón de habitantes, en comparación a la mayoría de los países en los que la tasa es cercana a 1 caso / millón de habitantes. La forma hereditaria alcanza el 38% del total de casos notificados y es debida a mutaciones en los codones 129, 118 y 200 del gen que sintetiza la PrP(1).
La ECJ se presenta en personas adultas con una edad media de aparición de 57 años (35-75), teniendo cinco formas de presentación: la forma clásica, la variante de Heidenhain, la forma atáxica, la variante con placas de Kuru y la vacuolar. La forma clásica de presentación corresponde al 70% de los diagnósticos y se caracteriza por una demencia progresiva de curso subagudo que se inicia con alteraciones conductuales y cognitivas, compromiso visual, alteraciones motoras, mioclonías y alteraciones del ciclo sueño vigilia. La enfermedad progresa invariablemente hacia la postración y el mutismo aquinético con una sobrevida promedio de 5,2 +/- 7 meses, sin que hasta la fecha se conozca un tratamiento efectivo para este mal.
Los exámenes complementarios son fundamentales en el diagnóstico de la forma clásica de la ECJ: el EEG muestra actividad epileptiforme pseudoperiódica en el 76,9% de los casos. En la gran mayoría de los pacientes la resonancia magnética cerebral muestra hiperintensidades en las secuencias T2 y difusión que afectan la corteza cerebral y ganglios basales. En el LCR de los pacientes se puede encontrar presencia de la proteína 14-3-3, sin ser éste un hallazgo patognomónico. La anatomía patólogica permite el diagnóstico definitivo, objetivando la presencia de espongiosis en putamen, caudado y corteza parietoccipital. La ECJ es una enfermedad de notificación obligatoria, cuyo diagnóstico en nuestro país generalmente se basa en los hallazgos clínicos y los exámenes complementarios. Lamentablemente en muy pocas oportunidades se cuenta con estudio de anatomía patológica post mortem(2).
Pese a que los trastornos de sueño están descritos en la ECJ, su presencia no ha sido cabalmente estudiada, probablemente por la preponderancia de los otros fenómenos clínicos. Esto contrasta con otra forma de enfermedad priónica llamada in-somnio fatal familiar (IFF), no descrita en Chile hasta la fecha. Se debe también a una mutación del gen de la PrP, tiene herencia autosómica dominante y cursa con insomnio severo asociado a sobreactividad motora, onirismo mental (estupor onírico), aumento de la actividad del sistema simpático y alteraciones del ciclo circadiano. La enfermedad progresa hacia una demencia y tiene una sobrevida de 4 a 72 meses, con edad de presentación entre los 40 y 70 años. A diferencia de la ECJ, en el IFF no se describen lesiones hiperintensas en la resonancia magnética cerebral, no hay actividad epileptiforme en el electroencefalograma, en la mayoría de los pacientes no se detecta proteína 14-3-3 en LCR y la anatomía patológica muestra una importante degeneración neuronal en los núcleos anteroventral y dorsomedial del tálamo con muy escaso compromiso cortical propio de otras encefalopatías espongiformes (ver Tabla 1).
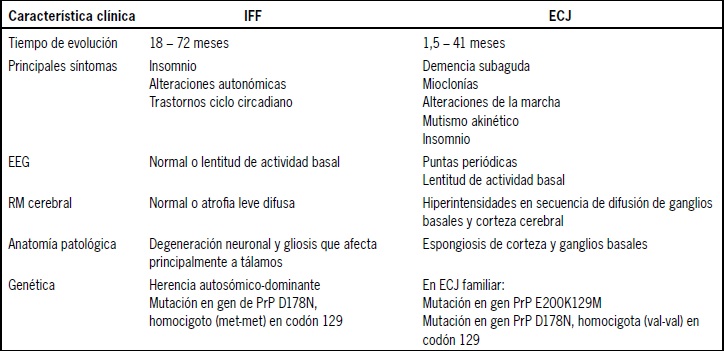
Tabla 1. Principales diferencias entre insomnio fatal familiar y enfermedad de Creutzfeldt-Jakob
La mutación descrita en el IFF es la misma de la ECJ familiar; sin embargo, las diferencias clínicas se deben a un polimorfismo en el codón 129 del gen que sintetiza la PrP, siendo homocigoto (Val-Val) en el IFF y heterocigoto (Val-Met) en la ECJ familiar.
CASOS CLÍNICOS
Se presentan 3 casos de pacientes que consultaron en el policlínico de Neurología del Hospital Clínico de la Universidad de Chile entre el año 2009 y 2010 con diagnóstico de ECJ en base a la anamnesis, el examen neurológico y los exámenes complementarios. En cada uno de los pacientes el insomnio fue un síntoma predominante al inicio de la enfermedad y se asoció a alucinaciones y alteraciones conductuales.
Caso 1
Paciente de 72 años de sexo femenino, soltera, trabajaba como comerciante. Sin antecedentes mórbidos relevantes en su historia personal salvo una dislipidemia leve sin terapia farmacológica. Consultó a Neurología en febrero del 2010 por un insomnio de conciliación y mantención progresivo de 5 meses de evolución, que no respondió a tratamiento con benzodiacepinas indicadas por su médico de cabecera. En la anamnesis no existían antecedentes psiquiátricos que explicaran la aparición del insomnio.
Pocas semanas antes de la consulta se agregan fallas leves de memoria, agitación psicomotora nocturna (gritaba en la noche y se levantaba frecuentemente de su cama), alucinaciones visuales, auditivas y delirio paranoídeo. Además un familiar notó inestabilidad de la marcha y temblor de extremidades.
El cuadro se asoció a baja de peso de 3 kg en un lapso de 3 meses, con apetito conservado.
En la evaluación neurológica de la primera consulta destacaba una paciente vigil, desorientada en fechas, inatenta, con fallas en evocación, apraxia ideomotora leve, acalculia y lenguaje hipofónico. En lo motor presenta temblor cinético de predominio izquierdo, reflejo plantar extensor a izquierda, sin paresias, reflejos osteotendineos normales. Sensibilidad conservada. Sin signos meníngeos.
Debido a que la paciente provenía de provincia, se decidió hospitalizarla para su estudio. Se realizó una resonancia magnética cerebral que mostró en secuencia de difusión signos involutivos, hiperintesidades en núcleo caudado y lenticular derecho, talámico medial bilateral y cortical frontal derecho (Figura 1). Todos los hallazgos con discreta representación en FLAIR y mapa ADC. El EEG en vigilia y somnolencia dentro de límites fisiológicos. Hemograma, perfil bioquímico y perfil tiroídeo normales. VDRL no reactivo. Nivel plasmático de vitamina B12 en rango normal. Punción lumbar: LCR claro, estudio citoquímico normal, Gram sin bacterias, tinta china negativa, VDRL no reactivo. Proteína 14-3-3 negativa.
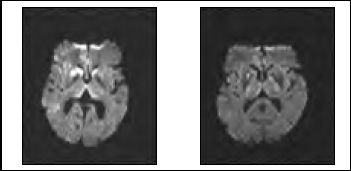
Figura 1. RM cerebral, en secuencia de difusión, de los casos N°1 y N°3 respectivamente, que muestran hiperintensidades anormales en lenticulares y tálamos.
Se realizó una polisomnografía convencional (Figuras 2 y 3) en la que destacaba una severa alteración en la arquitectura del sueño con muy baja eficiencia y severamente fragmentado por despertares durante toda la noche. Ausencia de sueño REM y un muy bajo porcentaje de sueño de “ondas lentas”. En el video se observó que en los despertares la paciente se mantenía en un estado estuporoso, similar al de vigilia, con movimientos no propositivos de extremidades superiores. No se registraron conductas violentas. Además en etapas NREM había ausencia de husos de sueño, complejos K y ondas de vértice. No se observó actividad epileptiforme. También se objetivó una elevación importante del índice de movimientos de piernas, con una alta proporción de eventos periódicos.
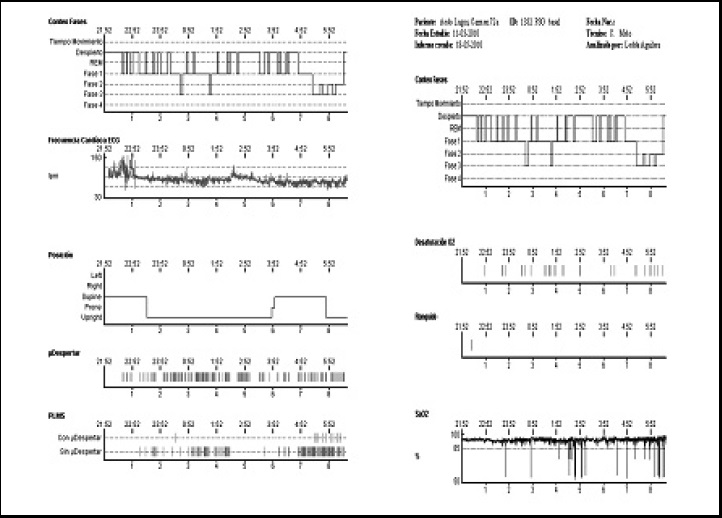
Figura 2. Hipnograma de la PSG realizada al caso N°1: destaca un sueño superficial interrumpido por numerosos despertares nocturnos, microdespertares espontáneos y movimientos periódicos de piernas.
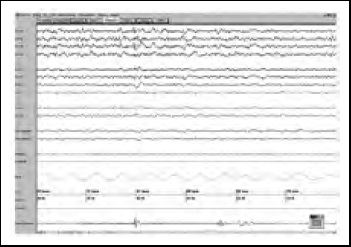
Figura 3. Caso N°1, visión de 120 segundos de polisomnografía en fase N1 de sueño. Se observan movimientos periódicos de piernas, parámetros respiratorios normales. Durante todo el examen no se visualizaron husos de sueño, complejos K y ondas de vértice.
Dos semanas después se repitió el EEG, destacando en vigilia leve lentitud focal en rango theta frontotemporal izquierda de naturaleza inespecífica. Nuevamente no se encontró actividad epileptiforme periódica.
La paciente fue dada de alta con diagnóstico de ECJ, falleciendo en su domicilio 10 meses después.
Caso 2
Paciente varón de 65 años, casado, guardia de seguridad, sin antecedentes mórbidos relevantes. Consultó en el policlínico de Neurología el año 2009 por un insomnio severo de seis meses de evolución, llegando a un estado de aparente vigilia continua nocturna, con mucha inquietud motora, y en ocasiones alucinaciones visuales complejas en las que el paciente veía personas en su habitación. Los últimos dos meses previos a la consulta el paciente tendía a aislarse socialmente, con menos iniciativa en las actividades de su vida diaria, presentaba fallas de memoria a corto y largo plazo y desconocía a algunos familiares. Progresivamente fue requiriendo más ayuda para vestirse, asearse y comer. Al insomnio se agregó alteración de la marcha, agravamiento general de sus déficits cognitivos e incontinencia de esfínteres.
Al examen neurológico destacaba: vigilia espontánea, lenguaje ininteligible (sonidos y monosílabos), no comprendía órdenes, trastorno severo de la atención. No lograba bipedestar. Espasticidad generalizada. Mioclonías espontáneas en distintos grupos musculares. La resonancia magnética cerebral mostró hiperseñal en secuencias T2 de sustancia gris periacueductal, subcortical frontal izquierda, peritrigonal derecha, putámenes, núcleos caudados y regiones dorsomediales del tálamo. El EEG mostró actividad epileptiforme periódica compatible con ECJ. Fue trasladado a otro centro hospitalario, falleciendo cinco meses después en su domicilio.
Caso 3
Paciente de sexo femenino, 58 años de edad, casada, con escolaridad básica completa, trabajaba como asesora del hogar. Dentro de los antecedentes mórbidos destacaba: hipotiroidismo subclínico por lo que se inició levotiroxina 100 mcg al día. En noviembre del año 2010 se hospitalizó en Psiquiatría con diagnóstico de episodio depresivo mayor y delirium, con una historia de 2 meses de evolución caracterizada por trastorno del ánimo, anhedonia, angustia, labilidad emocional, hipersomnia, disminución de apetito y baja de peso. Pocas semanas antes de la hospitalización la paciente comenzó a padecer de insomnio de conciliación y mantención muy severo, con alucinaciones visuales complejas durante la noche, estado de sobresalto permanente, angustia excesiva, delirio e ideas paranoides (presentía que ocurrirían desgracias como choques o asaltos).
Al examen mental impresionaba somnolienta, de ánimo depresivo, con motilidad disminuida, desorientación témporo-espacial, sólo comprendía órdenes de un elemento, apraxia ideomotora, acalculia. En el resto del examen neurológico destacaba: reflejos plantares flexores, parkinsonismo leve generalizado.
Se realizó un EEG que mostró una lentitud difusa de los ritmos de base con actividad epileptiforme lateralizada y periódica (PLEDS) enregión fronto-temporal derecha. TAC cerebral contrastada normal. Se realizó una resonancia magnética cerebral (Figura 1) que mostró en secuencia de difusión hiperseñales en la cabeza y cuerpo de ambos núcleos caudados, la mitad anterior del núcleo lenticular derecho e hiperseñal “en palo de jockey” en ambos tálamos. Múltiples focos de hiperintensidad en la corteza cerebral de predominio en región parietal derecha y fronto parietal mesial derecha. Los hallazgos descritos eran altamente compatibles con ECJ. LCR con citoquímico normal. Proteína 14-3-3 negativa. Se diagnóstico una ECJ esporádica, la paciente siguió con un deterioro cognitivo progresivo y falleció posteriormente en su domicilio.
DISCUSIÓN
Presentamos 3 pacientes cuyos antecedentes clínicos fueron compatibles con una EJC clásica, en ninguno de ellos se contó con confirmación histológica. En la historia de cada paciente destacaba un insomnio severo que apareció meses antes del diagnóstico definitivo. El trastorno de sueño fue progresivo y se asoció a alucinaciones visuales, sobreactividad motora y alteraciones conductuales. En uno de los casos se pudo realizar una polisomnografía en una etapa precoz de la enfermedad cuando no existía en el EEG actividad epileptiforme periódica. El resultado del examen evidenció una severa alteración de la arquitectura del sueño, destacando un sueño muy superficial y fragmentado por despertares y microdespertares, de baja eficiencia, y con ausencia de grafoelementos característicos de la etapa 2 de NREM. Se constató además la presencia de movimientos periódicos de piernas. Estos hallazgos son muy comunes de encontrar en el IFF y rara vez se han descrito en la ECJ(3,4).
Los pacientes durante el día presentaban alteraciones de su conducta que en algunos casos fueron catalogadas inicialmente como trastornos psiquiátricos, pero que progresaron en gravedad hasta instaurarse definitivamente una demencia. La aparición de mioclonias y alteraciones en la marcha fueron marcadores importantes del diagnóstico. Dentro de los exámenes complementarios fue fundamental encontrar hiperintensidades anormales en la resonancia magnética cerebral.
A diferencia de lo que ocurre en el IFF, los trastornos del sueño han sido poco estudiados en la ECJ. Cathala publicó una prevalencia de 39% de insomnio e hipersomnia en ECJ versus el 58% de controles hospitalizados. En casos de ECJ familiar se reconocen alteraciones de sueño en el 7% de los casos(5). En una serie chilena el insomnio se presentó en el 30% de los pacientes durante la fase prodrómica(6).
Morgado-Linares y col mencionan que 1 de 3 pacientes de sexo femenino, pertenecientes a una misma familia española con ECJ familiar confirmado con la mutación E200K, padeció dentro de su evolución clínica un insomnio severo(7).
Landolt publicó una serie de 7 pacientes con ECJ esporádica confirmada por anatomía patológica en la que todos los pacientes refirieron algún trastorno de sueño: 3 padecieron insomnio, 5 somnolencia diurna excesiva y 3 alucinaciones hipnagógicas. En la polisomnografía destacaban breves interrupciones de la vigilia por sueño NREM durante el día. Al igual que en nuestro caso, ningún paciente tenía husos de sueño ni actividad de vértice. Esta es una de las pocas publicaciones en las que se objetiva que las alteraciones del sueño, tanto clínicas como polisomnográficas, son muy frecuentes en los pacientes con ECJ esporádica y no específicas del IFF, destacando además que no se encontraron relaciones entre el insomnio y la afectación histológica de los núcleos dorsomediales y anteroventrales del tálamo(8).
Nuestros pacientes mostraban mucha actividad motora nocturna poco tiempo después de que aparecía el insomnio. Lugaresi fue el primero en acuñar el término agrypnia excitata (agrypnia = sin sueño) en referencia a un tipo de insomnio orgánico que cursa con sobreactividad motora, aumento de la actividad autonómica simpática y onirismo mental. Este tipo de insomnio se describe en el insomnio fatal familiar, el delirium tremens y el corea de Morvan (una forma de encefalitis autoinmune). En nuestros casos no se estudió la presencia de sobreactividad simpática(9).
Todos nuestros pacientes tenían alteraciones en la resonancia magnética cerebral típicas de la ECJ. En 2 de ellos las hiperintensidades de los lenticulares y tálamos eran predominantes por sobre las de la corteza. No existen evidencias publicadas acerca de una relación entre las alteraciones de la resonancia magnética cerebral y el insomnio en pacientes con ECJ. Meissner, en un estudio de 212 casos confirmados describió que las hiperintensidades típicas de ECJ se asociaban precozmente a demencia, mayor frecuencia de depresión y trastornos sensoriales. No se menciona al insomnio(10). En el IFF la resonancia magnética habitualmente es normal; sin embargo, los estudios con PET-scan describen alteraciones en el metabolismo del tálamo.
Si bien no se conocen exactamente los mecanismos responsables del insomnio en el IFF, se postula que la degeneración de los núcleos dorso medial y anteroventral del tálamo produce una desconexión funcional entre el núcleo reticular (generador de los husos de sueño) y estructuras del sistema límbico. Esto contrasta con los conceptos derivados de los primeros estudios realizados por Von-Economo en pacientes con encefalitis letárgica, quien atribuyó a las lesiones del hipotálamo anterior la presencia de insomnio y a las lesiones del hipotálamo posterior la presencia de somnolencia diurna excesiva(11).
Finalmente, pensamos que la descripción de nuestros casos, pese al sesgo de la falta de confirmación histológica, abre interesantes perspectivas para estudiar a futuro la relación entre afectación de tálamo
e insomnio en la ECJ.
REFERENCIAS
1. Cartier L, Quiroz G, Leiva M, Vergara C. Identificación clínica y patológica de las diversas formas de la enfermedad de Creutzfeldt Jakob en Chile. Rev Med Chile 2012;140:161-8.
2. Cartier L, Fernandez J, Ramirez E. Forma familiar de la enfermedad de Creutzfeldt- Jakob: marcadores genéticos en 4 familias chilenas. Rev Med Chile 2006;134:1116-22.
3. Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurology 2003;2:67–76.
4. Terzano MG, Parrino L, Pietrini V, Mancia D, Spaggiari MC, Rossi G et al. Precocious loss of physiological sleep in a case of Creutzfeldt Jakob disease: a serial polygraphic study. Sleep 1995;18:849–58.
5. Montagna P, Provini F. Prion disorders and sleep. Sleep Med Clin 2008;3:411–62.
6. Galvez S, Cartier L. Análisis clínico de una serie de 69 casos definitivos de Enfermedad de Creutzfeldt-jakob ocurridos en Chile entre 1960 y 1984. Rev Med Chile 1987;115:1148-54.
7 Morgado-Linares RY, J. Ruiz-Peña JL, Páramo MD, Díaz-Delgado M, Izquierdo G. Características clínicas de la enfermedad de Creutzfeldt-Jakob familiar y mutación E200K en España. Rev Neurol 2007;44:150-3.
8. Landolt HP, Glatzel M, Blattler T, Achermann P, Roth C, Mathis J et al. Sleepwake disturbances in sporadic Creutzfeldt-Jakob disease. Neurology 2006;66:1418–24.
9. Iriarte J, Ayuso T, Echavarri C, Alegre M, Urrestarazu E, Lacruz F et al. Agrypnia excitata in fatal familial insomnia. A video-poligraphic study. Neurology 2007;69:607-8.
10. Meissner B, Köhler K, Körtner K, Bartl M, Jastrow U, Mollenhauer U et al. Sporadic Creutzfeldt-Jakob disease: magnetic resonance imaging and clinical findings. Neurology 2004;63:450–6.
11. Nishino S. Hypothalamus, hypocretins/orexin, and vigilance control. En: P. Montagna and S. Chokroverty, Editors. Handbook of clinical neurology. Sleep disorders. Part 2. Vol 99 (3rd series). Elsevier B V, 2011;765-82.
Correspondencia:
 Dr. Mario Díaz Sepúlveda
Dr. Mario Díaz Sepúlveda
 Unidad de Epilepsia y Trastornos del Sueño Departamento de Neurología y Neurocirugía
Unidad de Epilepsia y Trastornos del Sueño Departamento de Neurología y Neurocirugía
 [email protected]
[email protected]
 562 2978 8262
562 2978 8262